
一种高纯度5-氟尿嘧啶合成工艺研究
2020-09-24 03:24陈小林
科技视界 2020年26期
陈小林
0 引言
5-氟尿嘧啶(5-FU)由Duschinsky 等[1]于1957 年首次合成,是一个重要的抗肿瘤药物,也是氟代嘧啶类抗肿瘤药物的重要中间体。目前氟尿嘧啶的合成方法(根据Scifinder 调研)可归纳为3 种方法:直接氟化法,缩合环合法及其他方法。直接氟化法是以尿嘧啶为原料,与惰性气体(N2)稀释的氟气或活性含氟化合物反应直接合成目标产物(5-氟尿嘧啶)[2-4]。此法工艺简单,收率高,但因氟气来源少,价格贵,氟气毒性大、对设备的要求高等原因,在产业化生产过程中应用较少。其他方法主要是以含氮杂环化合物的衍生物为起始原料,此法原料难得,成本高,不适宜产业化[5-6]。
目前主流工业化路线为缩合环合法,以氟乙酸甲酯为起始物料,经与甲酸乙酯在甲醇钠的催化下等到氟代丙醛酸甲酯烯醇式钠盐,此中间体与甲异脲(固体或者液体)环合成2-甲氧基-5-氟尿嘧啶,后经酸水解、重结晶等过程得到5-氟尿嘧啶[7-10]。此法路线长,效率低,但原料廉价(国内有大宗商品),反应条件温和(未见严苛的反应条件),易于产业化,总体成本低廉而得到国内生产厂家的青睐。

图1 高纯度5-氟尿嘧啶合成工艺反应路线示意图
作者以外购氟乙酸甲酯为起始物料,在甲醇钠的催化下与甲酸乙酯缩合得到烯醇式钠盐(中间体IV),不纯化直接与固体甲异脲硫酸盐环合成中间体V,然后在稀盐酸下水解得到粗品,粗品经过二甲亚砜、水2 步精制,得到HPLC 纯度>99.9%的高纯度成品。反应路线如图1 所示。
1 试验
1.1 试剂与仪器
上述合成路线中用到的试剂及仪器如下:
1.1.1 起始物料
1.1.2 其他物料
浓盐酸(HCl)-上海国药-AR。
1.1.3 溶剂
无水甲醇(CH3OH)-上海国药-AR。
纯化水(H2O)-自制-中国药典2015 年版。
1.1.4 辅助材料
药用活性炭C-上海长兴活性炭有限公司-药用级。
1.1.5 主要仪器及型号
天平(Sartorious-型号:BSA4202S-CW)。
真空烘箱(上海禾气玻璃仪器有限公司:DZF-6020)。
带加热磁力搅拌器(上海禾气玻璃仪器有限公司:DF-101S)。
隔膜真空泵(上海禾气玻璃仪器有限公司:M22CHT)。
高压液相仪(water-型号:e2695-2998PDA)。
液质联用仪(Q-Tofmicro-型号:Q-Tofmicro)。
核磁共振波谱仪(BRUKER-型号:BRUKER-400)
1.2 合成方法
1.2.1 氟代丙醛酸甲酯烯醇式钠盐(化合物IV)的合成
(1)投料物料配比。
(2)合成步骤。
往干燥的三口反应瓶(3L)内加入甲苯,开始搅拌,氮气置换三次;继续往反应瓶内加入固体甲醇钠,然后内温降至0~15℃;往反应瓶内继续滴加甲酸乙酯液体,控制内温0~20℃,1 h 内滴加完毕;滴加氟乙酸甲酯液体,控制内温不超过30℃,1 h 内滴完;继续搅拌升温至30~40℃,保温反应9 h。此反应液无须处理直接进行下一步。

表1 化合物IV 投料物料配比表
1.2.2 2-甲氧基-5-氟尿嘧啶(中间体V)的合成
(1)投料物料配比。
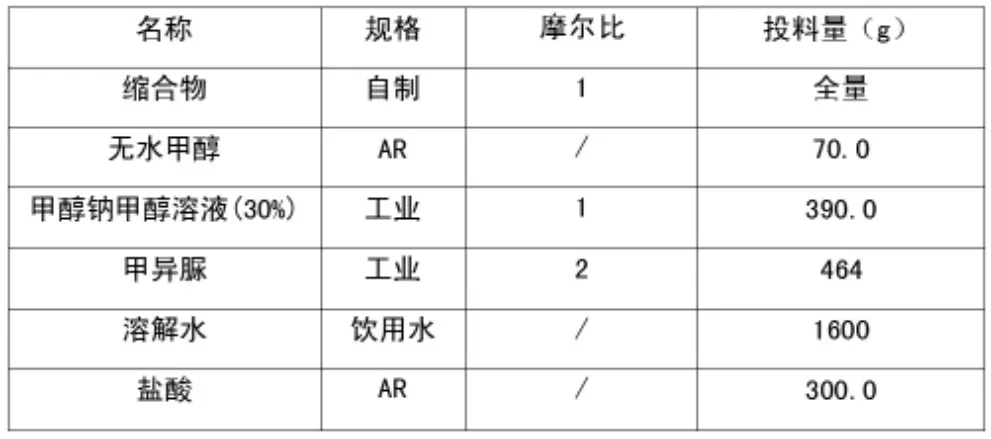
表2 中间体V 投料物料配比表
(2)合成步骤。
上述反应液转入另一个5 L 的三口瓶中,氮气置换三次,将瓶内温度冷却至10℃,开启搅拌,依次加入甲醇钠溶液、固体甲异脲,控制内温35~45℃。保温搅拌9 h 后,取样送中控,检测含量,若反应液中产物含量≥10.0%,则终止反应,进行下一步操作。若<10.0%,则继续反应,并每隔3 小时取样直至含量≥10.0%。
后处理:加入水1.6 L,搅拌30 min 使反应液溶清,停止搅拌,静置分层,将下层水相转入至另外一个反应瓶中内。水相冷却至20~35℃,滴加浓盐酸,调节pH 至3~4,析出大量淡黄色固体,降温至10℃,继续搅拌30min 后过滤。滤饼折干后约190.5 g,摩尔收率:60.6%。MS ESI+=145.10(M+1);1H-NMR(400 MHZ,DMSO)δ∶3.863(s,3H),7.852-7.861(d,J=3.6,1H),12.853(s,1H)。
1.2.3 粗品(VI)的合成(1)投料物料配比。

表3 粗品(VI)投料物料配比表
(2)合成步骤。
往三口瓶(3 L)中加入纯化水(200 ml),搅拌下加入2-甲氧基-5-氟尿嘧啶化合物;氮气置换3 次后开始滴加盐酸溶液(4M)),1 h 左右滴完;内温升至50~60℃,保温反应3 h;取样送检(HPLC 中控),当液相结果中环合物含量≤5%(面积归一法)时,反应结束后,内温冷却至5~15℃,过滤,得粗品,粗品折干重月116.2 g,摩尔收率:91.9%。
1.2.4 粗品精制
1)投料物料配比。

表4 粗品精致投料物料配比表
2)合成步骤。
(1)二甲亚砜精制。
将DMSO 和粗品加入单口反应瓶内,搅拌升温至内温70~80℃,反应液溶清,保温搅拌1h;开始降温至内温降至20~25℃,保温搅拌3h 后过滤,得滤饼,滤饼无须干燥直接投下一步。
(2)水精制
将二甲亚砜精制得到的粗品加入纯化水中,搅拌升温至回流,反应液溶清,加入活性炭;回流保温30mins,趁热过滤,滤液缓慢降温至T=10℃,析晶2 小时后过滤,滤饼50℃真空干燥16 h后,得成品64.3 g,摩尔总收率:71.4%。m.p.284.1-284.4(药典:282-286℃)。MS ESI+=131.10 (M+1),153(M+Na);1H-NMR(400MHZ,DMSO)δ∶7.721-7.736(d,J=6.0,1H),10.702(s,1H),11.489(s,1H)。
2 结果与讨论
2.1 2-甲氧基-5-氟尿嘧啶(中间体V)的条件优化
2.1.1 缩合反应
由于化合物IV 是烯醇式钠盐,目前没有很好的分析方法来监控,故这步优化后直接做成中间体V,优化指标主要是通过中间体V 的收率及纯度来衡量。主要优化参数为:
碱的种类(甲醇钠、氢氧化钠、三乙胺)、碱当量(1.5eq、2.0eq、2.2eq、2.5eq)、缩合反应温度(25±5℃、35±5℃、50±5℃)、甲酸乙酯当量(2eq、2.25eq、3.0eq)、缩合反应时间(4 h、9 h、16 h),确定这步最佳反应条件为:甲醇钠(2.2eq)、反应温度(35±5℃)、甲酸乙酯用量(2.25eq)、反应时间(9 h)。
2.1.2 环合反应
环合反应可以通过高压液相来监控,通过生成中间体V 的含量及纯度两个指标来优化各参数。具体优化参数如下:
环合反应温度(25±5℃、40±5℃、50±5℃)、环合反应甲醇钠当量(0.7eq、1eq、1.3eq)、固体甲异尿当量(1eq、2eq、3eq),确定最优工艺参数为:环合温度(40±5℃)、环合碱当量(1.3eq)、固体甲异尿当量(2eq)。
此两步摩尔总收率:60.6%,中间体V 的HPLC 纯度为95.0%以上。
2.2 粗品(VI)的条件优化
此步水解反应主要考察不同的酸水解(浓盐酸、硫酸、乙酸),溶剂体积(10 V、5 V、3 V),反应温度(35±5℃、45±5℃、55±5℃)等工艺参数。
确定最佳工艺参数为:水解酸用浓盐酸,水体积为5 V,反应温度为55±5℃。
这步反应摩尔收率:91.9%。
2.3 精制的条件优化
第一次精制溶剂筛选(DMF、DMSO、H2O、丙酮),溶剂体积筛选(3 V、5 V、10 V);第二步精制溶剂主要筛选溶剂体积(5 V、10 V、15 V),药用活性炭用量(5%、10%、15%)。
确定最佳工艺参数:第一步精制溶剂选5 V 的二甲亚砜重结晶;第二步精制溶剂选10 V 的水,活性炭选10%的量。两步重结晶摩尔收率为:71.4%,HPLC 纯度>99.9%。
3 总结
通过对本路线各中间体步骤的优化,总路线的摩尔收率39.8%,HPLC 纯度99.9%以上(面积归一法),整条反应路线未使用到特殊反应条件及试剂,适合工业化,且通过此路线得到的产品质量超中国药典及欧洲药典标准。
猜你喜欢
当代化工研究(2023年16期)2023-09-11
世界农药(2023年8期)2023-09-04
中国药学药品知识仓库(2022年10期)2022-05-29
中国烟草学报(2021年4期)2021-09-26
水泵技术(2021年4期)2021-01-22
汕头大学学报(自然科学版)(2020年4期)2020-12-14
山东煤炭科技(2018年1期)2018-12-05
食品与机械(2018年5期)2018-07-14
橡胶工业(2016年2期)2016-02-23
医学美学美容·中旬刊(2015年2期)2015-10-21
