
固相萃取-衍生化-气相色谱-质谱联用法同时测定尿液中4种阿片类物质
2020-09-23 06:58刘雅珣倪浏阳
色谱 2020年11期
高 吭, 刘雅珣, 柯 威, 刘 凯, 倪浏阳, 陶 涛
(1. 湖北省黄石市公安局, 湖北 黄石 435000; 2. 黄石市公安司法鉴定中心, 湖北 黄石 435000; 3. 黄石市毒品检验鉴定中心, 湖北 黄石 435000)
海洛因被联合国列为一类管制毒品,也被中国列管为主要毒品之一。目前,我国240.4万名吸毒人员中,海洛因滥用者为88.9万名,占37%[1]。海洛因在人体内通过脱乙酰化作用迅速代谢,生成O6-单乙酰吗啡(O6-acetylmorphine),然后再缓慢地代谢为吗啡(morphine)。公安机关使用胶体金尿检板检测尿液中的吗啡[2,3]和O6-单乙酰吗啡,作为吸食海洛因的判断依据。而服用含吗啡、可待因(codeine)和乙酰可待因(acetyl codeine)的镇咳药[4,5],同样会使吗啡胶体金尿检板呈阳性。
检测尿液中吗啡、O6-单乙酰吗啡的方法主要有液相色谱-质谱联用法[6-8]和气相色谱-质谱联用法[9-12]。液相色谱-质谱联用法具有样品前处理简单、检测速度快和灵敏度高的优点,缺点是价格昂贵、使用难度大、维护成本高、普及率低。高普及率的气相色谱-质谱联用仪与液相色谱-质谱联用仪相比更符合实际工作的需要。董镧等[11]报道利用液-液提取和丙酸酐衍生化法检测生物样品中的吗啡,回收率在83.2%~87.4%之间,检出限为0.02 μg/mL;何国标等[12]报道了GDX403、C18和HLB 3种固相萃取柱对尿液中吗啡、可待因、乙酰可待因和O6-单乙酰吗啡的萃取效果,以HLB柱最优,回收率在79%~85%之间,其中吗啡检出限为0.05 μg/mL。
吗啡、O6-单乙酰吗啡、可待因和乙酰可待因在酸性条件下以阳离子状态存在。本文选用MCX混合型阳离子交换固相萃取柱[13-16]萃取尿液中4种阿片类物质,再用N-甲基-N-(三甲基硅烷基)三氟乙酰胺(MSTFA)[17,18]作为衍生化试剂对吗啡、O6-单乙酰吗啡和可待因进行衍生,最后用气相色谱-质谱联用法测定。该方法简单、快速、灵敏、准确、可批量操作,适合尿液中4种阿片类物质的快速检测与定量分析。
1 实验部分
1.1 仪器、试剂与材料
AutoSPE-06plus六通道全自动固相萃取仪(厦门睿科仪器有限公司); Auto EVA 30位氮吹浓缩仪(厦门睿科仪器有限公司); 7890BGC-5977BMSD气相色谱-质谱联用仪(美国Agilent公司); Purelab Option Q7超纯水仪(英国ELGA公司); FD115干燥箱(德国IRM公司); TOLEDO FE20K pH计(美国METTLER公司); OASIS MCX 3cc LP Extraction Cartridge固相萃取柱(美国Waters公司)。
无水磷酸二氢钠、无水磷酸氢二钠(分析纯,国药集团化学试剂有限公司);氨水、甲酸(分析纯,天津市天力化学试剂有限公司);甲醇(色谱纯,西陇化工股份有限公司);乙酸乙酯(色谱纯,美国TEDIA公司);所用水为超纯水,由Purelab Option Q7超纯水仪制备。
衍生化试剂MSTFA(美国Sigma-Aldrich公司)。标准品:吗啡、O6-单乙酰吗啡、可待因、乙酰可待因(1 mg/mL,美国Cerilliant公司)。
1.2 标准溶液的配制
分别移取4种阿片类物质标准品100 μL,置于1 mL容量瓶中,用甲醇稀释并定容,混匀,配制得100 μg/mL的阿片类物质混合标准溶液,于4 ℃冰箱保存待用;用甲醇稀释,配制0.02、0.1、0.2、0.4、0.8、1、10、20 μg/mL的混合标准溶液。
1.3 实验方法
1.3.1样品的前处理
取待检尿液5 mL,添加0.2 mol/L pH=6的磷酸盐缓冲液(PBS)5 mL,混匀,离心,上清液待全自动固相萃取仪处理。
1.3.2固相萃取和衍生化
将MCX固相萃取柱安装于全自动固相萃取仪上,固相萃取仪条件见表1。
洗脱液在60 ℃干燥氮气流下浓缩至干,吹干后的残渣置于105 ℃干燥箱内干燥0.5 h,冷却至室温后取出,加入0.1 mL乙酸乙酯溶解残渣,添加MSTFA 20 μL,混匀,密闭,微波炉功率385 W加热2 min,液体转移至样品瓶内,上机检测。
1.3.3色谱条件
色谱柱:HP-5MS色谱柱(30 m×0.25 mm×0.25 μm,美国Agilent公司);进样口温度:280 ℃;进样方式:不分流进样;载气:高纯氦气(99.999%);流速:1 mL/min。升温程序:初始温度150 ℃,保持1 min,以10 ℃/min速率升至280 ℃,保持10 min。后运行温度:300 ℃;后运行时间:3 min;传输线温度:280 ℃。
1.3.4质谱条件
离子源温度:230 ℃;四极杆温度:150 ℃;采集类型:全扫描;质量数范围:35~500 amu;溶剂延迟时间:4 min。可待因、吗啡和O6-单乙酰吗啡的衍生化产物分别为:O6-三甲基硅烷基可待因(codeine, TMS derivative),O3,O6-双(三甲基硅烷基)吗啡(morphine, 2TMS derivative)和O3-三甲基硅烷基-O6-乙酰基吗啡(O6-acetylmorphine, TMS derivative),它们和乙酰可待因的保留时间、定性离子和定量离子见表2,质谱图见图1。

表 1 尿液样品的固相萃取条件

表 2 4种分析物的保留时间、定性离子和定量离子

图 1 (a)O6-三甲基硅烷基可待因、(b)O3,O6-双(三甲基 硅烷基)吗啡、(c)O3-三甲基硅烷基-O6-乙酰基吗啡和(d)乙酰可待因的质谱图 Fig. 1 Mass spectra of (a) codeine, TMS derivative, (b) morphine, 2TMS derivative, (c) O6-acetylmorphine, TMS derivative, and (d) acetyl codeine
2 结果与讨论
2.1 上样和洗脱流速的选择
本实验分别考察了上样和洗脱流速为0.5, 1.0, 2.0, 3.0 mL/min时4种阿片类物质的回收率。如图2所示,随着上样流速升高,4种阿片类物质的回收率随之下降,但选择1.0 mL/min上样流速时,回收率较0.5 mL/min时下降不明显,因上样总量加全自动固相萃取仪上样死体积共计12 mL,为提高萃取效率,选择1.0 mL/min的上样流速。如图3所示,选择0.5 mL/min的洗脱流速时,洗脱液对固相萃取柱保留的阿片类物质洗脱强度不足,回收率较低。当洗脱流速大于等于1.0 mL/min时,回收率接近100%,且流速为1.0 mL/min时,误差较低,因洗脱液仅2.5 mL,加快洗脱流速并不会对萃取效率带来明显提升,综合考虑,选择1.0 mL/min的洗脱流速。

图 2 不同上样流速对尿液中4种阿片类物质回收率的影响(n=5)Fig. 2 Effect of loading flow rate on the recoveries of the four opioids in urine (n=5)

图 3 不同洗脱流速对尿液中4种阿片类物质回收率的影响(n=5)Fig. 3 Effect of elution flow rate on the recoveries of the four opioids in urine (n=5)
2.2 淋洗液中甲酸体积分数的选择
MCX填料可克服传统硅胶基质混合型固相萃取吸附剂的局限性,具备离子交换和反相双重保留模式。尿液中阿片类物质在pH=6时呈阳离子状态,与MCX填料中的磺酸基相互作用得以保留,同时非极性及弱极性杂质也保留在填料中的聚苯乙烯/二乙烯苯基体上。使用甲酸甲醇溶液淋洗固相萃取柱,可降低非极性及弱极性杂质的干扰。本实验考察了3种不同体积分数的甲酸甲醇淋洗液对回收率的影响,实验结果如图4所示,选择3%(v/v)甲酸甲醇溶液淋洗去除杂质时,回收率较高。

图 4 不同体积分数的甲酸甲醇淋洗液对尿液中4种阿片类物质回收率的影响(n=5)Fig. 4 Effect of volume percentage of formic acid in methanol washing solvent on the recoveries of the four opioids in urine (n=5)
2.3 淋洗液体积的选择
用甲酸甲醇溶液淋洗杂质,会对保留在固相萃取柱上的阿片类物质造成一定损失。本实验考察了3种不同淋洗体积对回收率的影响,实验结果如图5所示。选择1 mL 3%(v/v)甲酸甲醇溶液淋洗时,回收率较好。不使用3%(v/v)甲酸甲醇溶液淋洗的情况下,目标物受基质影响,回收率的离散程度较大。而随着3%(v/v)甲酸甲醇溶液使用量增加至2 mL,阿片类物质的损失随之增加,回收率下降。

图 5 不同体积的淋洗液对尿液中4种阿片类物质回收率的影响(n=5)Fig. 5 Effect of volume of washing solvent on the recoveries of the four opioids in urine (n=5)

图 6 不同体积分数的氨水甲醇洗脱液对尿液中4种阿片类物质回收率的影响(n=5)Fig. 6 Effect of volume percentage of ammonium hydroxide in methanol eluent solvent on the recoveries of the four opioids in urine (n=5)
2.4 洗脱液中氨水体积分数的选择
4种待测阿片类物质在洗脱液中呈游离碱或阴离子状态,失去与磺酸基的相互作用。本实验考察了3种不同体积分数的氨水甲醇溶液作为洗脱液对回收率的影响,实验结果如图6所示。使用3%(v/v)氨水甲醇溶液作为洗脱液时,回收率较差,当洗脱液中氨水的体积分数大于5%时,回收率随着氨水体积分数的上升变化不明显。确定选择5%(v/v)氨水甲醇溶液作为洗脱液。
2.5 固相萃取柱吹干时间的选择
在固相萃取的操作中,为防止出现洗脱液和淋洗液不混溶时导致的乳化现象,会在洗脱前将固相萃取柱吹干。部分型号的固相萃取填料如C18等,如不吹干或吹得过干,回收率都会受到较大的影响,本实验考察了使用0.1 MPa压力的干燥氮气流,在5种不同时长(1、3、5、8、10 min)下吹干固相萃取柱对回收率的影响。实验结果如图7所示,MCX固相萃取柱在不同的干燥程度下,回收率没有明显变化。为提高萃取效率,选择将固相萃取柱吹干1 min。
2.6 固相萃取净化效果
在优化的固相萃取条件下,空白尿液中标准添加水平为0.2 μg/mL时,样品的总离子流图中4种分析物完全分离,峰形良好,基本消除了共流出的基质杂质(见图8)。

图 7 不同吹干时间对尿液中4种阿片类物质回收率的影响(n=5)Fig. 7 Effect of cartridge drying time on the recoveries of the four opioids in urine (n=5)

图 8 0.2 μg/mL加标的空白尿液经固相萃取净化后的总离子流色谱图Fig. 8 Total ion current chromatogram of blank urine spiked at 0.2 μg/mL purified by solid phase extraction
2.7 线性范围、检出限和定量限
将1 mL的系列标准工作液(0.02、0.1、0.2、0.4、0.8 μg/mL)浓缩至干,在确定的实验条件下检测,以定量离子峰面积(y)对其在标准工作液中的质量浓度(x, μg/mL)进行线性回归,绘制校准曲线,得到线性方程和相关系数(r2)。结果显示,4种分析物的线性关系良好,r2均大于0.998。按本实验方法对0.02 μg/mL的空白加标尿液进行检测,分别以3倍和10倍信噪比(S/N)确定检出限(LOD)和定量限(LOQ),结果见表3。

表 3 4种分析物的线性范围、线性方程、相关系数、检出限和定量限
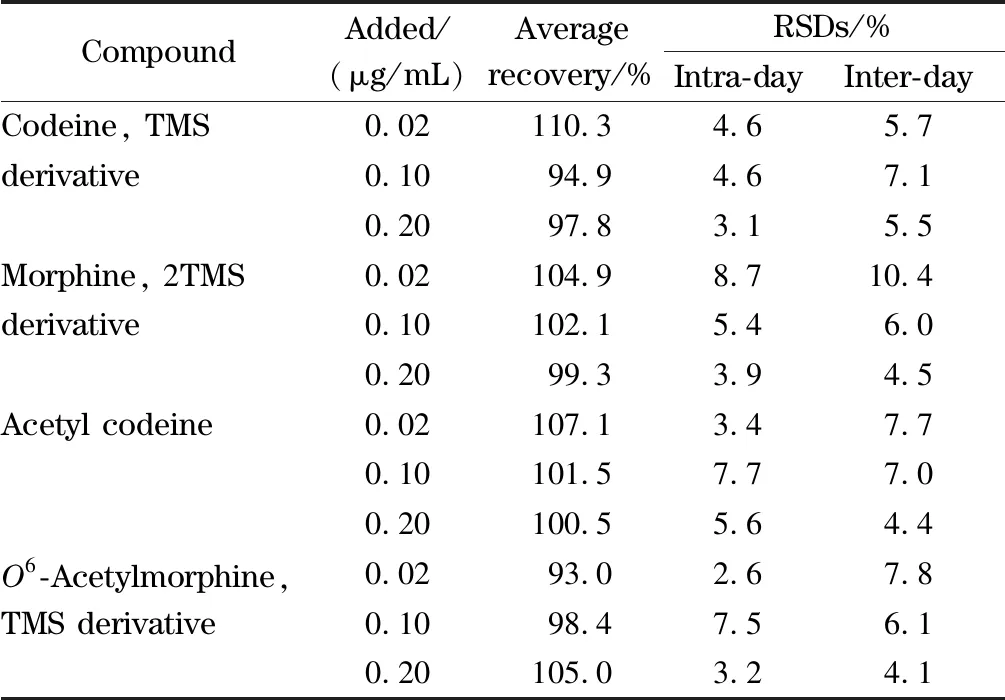
表 4 加标尿样中4种阿片类物质的回收率、日内精密度(n=5)
2.8 回收率和精密度
根据定量限及实际样品中的含量范围,在空白尿液中添加0.02、0.1、0.2 μg/mL 3个水平的阿片类物质。按照已确定的方法,每个样品平行检测5次,考察日内精密度;连续检测3天,考察日间精密度。如表4所示,4种分析物的平均回收率为93.0%~110.3%,日内精密度为2.6%~8.7%,日间精密度为4.1%~10.4%。
2.9 实际样品的检测
应用本方法对服用过复方甘草片的志愿者尿样进行检测,该尿样检出吗啡和可待因,其含量分别为0.091 5和0.012 6 μg/mL。
3 结论
本文建立了固相萃取-衍生化-气相色谱-质谱联用同时测定尿液中吗啡、O6-单乙酰吗啡、可待因和乙酰可待因的方法。本方法样品前处理采用MCX固相萃取柱萃取尿液中阿片类物质,用MSTFA三甲基硅烷化。结果表明,方法的线性关系良好,回收率和灵敏度高,具有定性定量准确、快速的特点,可以满足公安机关实际工作需要。
猜你喜欢
分子催化(2022年1期)2022-11-02
化工自动化及仪表(2021年3期)2021-06-04
食品工程(2020年4期)2021-01-20
中国油脂(2020年3期)2020-04-10
酿酒科技(2019年10期)2019-11-12
时代英语·高一(2018年5期)2018-11-19
时代英语·高一(2017年5期)2017-11-14
云南中医学院学报(2015年2期)2015-07-31
质谱学报(2015年5期)2015-03-01
中国塑料(2014年4期)2014-10-17
