
加压毛细管电色谱法高效分离分析葛根多糖中的单糖
2020-09-23 06:58许歆瑶SoumiaCHEDDAH
色谱 2020年11期
许歆瑶, Soumia CHEDDAH, 王 彦, 阎 超
(上海交通大学药学院, 上海 200240)
葛根为豆科植物野葛PuerariaLobate(Willd.)ohwi的干燥根,其产地较广,资源丰富,可分为粉葛(PuerariathomsoniiBenth)和柴葛(PuerarialobataOhwi)两种[1,2],粉葛为豆科植物甘葛藤的干燥根;柴葛习称野葛,为豆科植物野葛的干燥根。富含异黄酮类、多糖类、氨基酸及微量元素等物质,具有营养保健功效和突出的药用价值,故享有“南方人参”的美誉,其有效成分在维持心血管系统稳定性、保护脑神经、抗氧化、防止肝肾损伤、改善代谢与免疫功能等方面均有一定的药理作用[3-5]。但目前大多研究主要关注葛根素及总黄酮成分,对葛根多糖研究较少[6,7]。糖类物质由于没有紫外吸收,并且具有很强的极性,所以难以直接分离检测。近年来对植物多糖中单糖分析检测的方法主要包括衍生化的液相色谱法[8,9]、亲水作用色谱[10]、气相色谱-质谱联用[11,12]、液相色谱-质谱联用[13-15]及近年发展起来的离子色谱法[16,17]等,对枸杞多糖、葛根多糖、鹿茸多糖等中药多糖的单糖组成进行了测定,这些方法存在衍生操作较为繁琐、分离柱效欠佳、质谱检测器价格高昂等不足。多糖中单糖组成及含量不同可能导致多糖空间结构不同,从而导致生物活性不同,因此,对葛根多糖进行提取并测定葛根多糖的单糖组成对其活性分析和质量控制具有重要意义。
加压毛细管电色谱(pressurized capillary electrochromatography, pCEC)是一种结合了毛细管电泳(capillary electrophoresis, CE)和毛细管液相色谱(capillary liquid chromatography, cLC)优点的高效电动微分离技术。pCEC兼具CE和cLC的双重分离机理,既可以分离中性物质也可分离带电物质,同时又具备高柱效、高分辨率、高选择性和快速分离等优点[18]。目前,尚未有利用加压毛细管电色谱法同时分离葡萄糖(glucose, Glc)、鼠李糖(rhamnose, Rha)、甘露糖(mannose, Man)、阿拉伯糖(arabinose, Ara)、半乳糖(galactose, Gal)、核糖(ribose, Rib)、木糖(xylose, Xyl)和岩藻糖(fucose, Fuc) 8种中性单糖并应用于葛根多糖单糖组成鉴定的报道。
本研究利用提取时间短、提取效率高、操作简便的超声提取法提取葛根多糖,并利用响应面分析法对提取条件进行探索优化;利用改善的1-苯基-3-甲基-5-吡唑啉酮(PMP)衍生法,建立加压毛细管电色谱-紫外检测器新方法探索多种中性单糖的分离,优化分离条件,实现单糖的快速高效分离检测,并对中药葛根实际样品的单糖组成成分进行探究与分析,为单糖化合物的分离检测提供新方法并为葛根多糖的单糖组成提供参考。
1 实验部分
1.1 仪器、试剂与材料
TriSepTM2100加压毛细管电色谱仪,包括柱上紫外检测器、微流控系统、溶剂输送系统、高压电源和数据采集系统(美国Unimicro Technologies公司); KQ5200DE型超声波清洗器(昆山超声仪器有限公司); SHZ-Ⅲ型循环水真空泵(上海大颜仪器设备有限公司); RE-52AA旋转蒸发器(上海亚荣生化仪器厂); DF-101S集热式恒温加热磁力搅拌器(巩义市予华仪器有限责任公司); Milli-Q纯净水仪(德国默克密理博公司)。
D(+)-岩藻糖(纯度>98%)、1-苯基-3-甲基-5-吡唑啉酮(纯度>99%)购自上海阿拉丁生化科技股份有限公司;D(+)-甘露糖(纯度>99%)、D(+)半乳糖(纯度>98%)购自Alfa Aesar(阿法埃莎)中国化学有限公司;D(+)葡萄糖分析对照品(纯度>99%)购自上海麦克林生化科技有限公司;D(+)木糖(纯度>98%)购自上海迪柏生物科技有限公司;L(+)鼠李糖(纯度>98%)购自上海迈瑞尔化学技术有限公司;D-阿拉伯糖(纯度>98%)购自上海易恩化学技术有限公司;D-核糖(纯度>99%)购自萨恩化学技术(上海)有限公司。乙腈、氨水、乙酸、三氯甲烷、三氟乙酸(TFA)、石油醚、无水乙醇等均为国产分析纯,购自国药集团化学试剂有限公司。粉葛根饮片购于河北纽恩堂有限公司(药材原产地:湖南),柴葛根茶饮片购于神农金康公司(药材原产地:湖南)。
1.2 标准单糖的衍生化
1.2.1试剂配制
衍生化试剂:精密称取PMP 0.870 g,置于10 mL容量瓶中,加入甲醇溶解并定容,摇匀,制得0.5 mol/L的衍生化试剂PMP甲醇溶液,保存于4 ℃冰箱备用。
标准单糖混合溶液:准确称取甘露糖、鼠李糖、核糖、葡萄糖、半乳糖、木糖、阿拉伯糖、岩藻糖标准品各10 mg于5 mL容量瓶中,加氨水溶解并定容,摇匀,制得2 g/L的8种单糖对照品碱性混合溶液,保存于4 ℃冰箱备用。
1.2.2标准单糖混合物的衍生化
取200 μL 2 g/L 8种单糖混合溶液于反应管中,加入200 μL 0.5 mol/L的PMP甲醇溶液,封盖,在70 ℃反应100 min,反应结束置室温冷却后,真空干燥除氨,再加入2 mL三氯甲烷,充分振荡,于3 000 r/min离心5 min,弃去有机相,除去过量的衍生试剂,重复萃取操作3次后,取水相过0.22 μm滤膜后适当稀释,待进样分析。
1.3 样品制备
1.3.1葛根多糖的超声提取
称取适量葛根样品加5倍量石油醚在80 ℃下回流2 h,脱脂后过滤,干燥备用[19];称取每份10 g葛根样品,按照一定液料比(粉葛多糖液料比20 mL/g,柴葛多糖液料比40 mL/g)、超声温度90 ℃、超声时间30 min、超声功率180 W进行超声提取;抽滤,滤液浓缩后,加入l/5体积Sevage试剂(氯仿-正丁醇,体积比为4∶1)除蛋白,后加入4倍体积的无水乙醇使多糖沉淀,4 ℃静置过夜,抽滤得沉淀,真空干燥后得到葛根多糖[20,21]。
1.3.2葛根多糖的水解
准确称取超声提取后的粉葛和柴葛多糖样品各5 mg置于圆底烧瓶中,加入2 mol/L TFA溶液2 mL, 120 ℃条件下水解2 h,冷却后加甲醇旋干除去TFA,重复3次至中性,加入1 mL超纯水溶解后得到多糖水解液。
1.3.3葛根多糖水解样品衍生
取2种多糖水解液各200 μL于反应管中,加入200 μL 0.5 mol/L的PMP甲醇溶液和800 μL氨水,同1.2.2节衍生方法进行PMP衍生化和后续处理,得到多糖水解衍生物,过0.22 μm滤膜后,待进样分析。
1.4 pCEC-UV分离单糖色谱条件
色谱柱:C18核壳型Halo柱(20 cm×100 μm, 2.7 μm,苏州环球色谱有限责任公司);流动相为乙腈-50 mmol/L pH 4.1的醋酸铵水溶液(18∶82, v/v);流速0.1 mL/min;施加电压-20 kV;柱温30 ℃;检测波长:250 nm;进样量5 μL;毛细管电色谱实际分流比1∶400。
1.5 提取工艺响应面法优化实验
利用Design-Expert 8.0软件,采用Box-Behnken设计实验方案,以液料比(A)、超声时间(B)、超声温度(C)和超声功率(D)为考察变量,以葛根多糖粗提率为响应值,设计四因素三水平响应面分析试验,试验因素水平见表1。

表 1 Box-Behnken设计试验的因素水平
2 结果与讨论
2.1 pCEC分离条件的探索优化
选用8种单糖衍生物为分离对象,以pCEC-UV分离模式为平台,探究优化的衍生方法、缓冲盐浓度、缓冲盐pH、施加电压、毛细管色谱柱填料及流动相比例对分离单糖衍生物的影响。
2.1.1衍生化方法优化
依据传统方法[22-24],通常用NaOH提供衍生反应的碱性条件,反应过后需加入HCl进行中和。本研究利用该法进行的前期试验表明,由于毛细管电色谱分离时,毛细管内部容积小至几微升,分离易受样品基质轻微变化的影响,传统方法处理所得样品含有较高浓度NaCl(通常为0.3 mol/L),易造成毛细管入口处洗脱液组成的不规则变化[25],实验中发现会导致毛细管柱堵塞、加电困难,从而使分离效果不佳且柱效低。后尝试将单糖直接溶解在氨水中,反应后真空干燥除氨,从而解决传统衍生方法中NaCl对pCEC分离单糖的影响并简化了操作。
2.1.2缓冲盐浓度对分离的影响
首先用3 μm C18柱在加压-22 kV、缓冲盐pH 4.9条件下,用10、25、50、100 mmol/L的醋酸铵缓冲液作为水相对8种单糖衍生物进行分离。结果表明,缓冲盐浓度对物质的保留时间有较大影响,缓冲盐浓度较低时,物质电泳效果较差,保留时间长达近1 h,且核糖与鼠李糖、阿拉伯糖与木糖无法分离。当浓度达到50 mmol/L时,分离时间缩短到20 min内,且核糖与鼠李糖实现完全分离,阿拉伯糖与木糖分离度增加。当浓度为100 mmol/L时,由于产生的电流过大,无法形成稳定的基线。故选择50 mmol/L的缓冲液。
2.1.3缓冲盐pH对分离的影响
在加压-22 kV、缓冲盐浓度50 mmol/L条件下,用pH 5.5、4.9、4.1和3.7的醋酸铵缓冲液对8种单糖衍生物进行分离。结果表明,当pH降低时,由于在酸性条件下物质带电及电泳效果增强,保留时间缩短,阿拉伯糖和木糖分离度增加,葡萄糖和半乳糖分离度降低,其余分离度无显著变化。根据分离情况选择pH 4.1。
2.1.4施加电压对分离的影响
分别在cLC (0 kV)和pCEC (-15 kV到-22 kV)模式下对8种单糖衍生物进行分离。结果表明,cLC条件下约100 min全部出峰,pCEC -22 kV时可将分离时间缩短到20 min内,相比传统LC方法对单糖的分离[23,24,26,27],显著缩短了分离时间;且柱塞型电渗流驱动力和抛物线型压力流驱动力联合驱动样品的分离,大大提高柱效及分离度,不加电时无法分离的核糖和鼠李糖在-20 kV时达到完全分离。综合考虑,选择-20 kV为分离电压。
2.1.5色谱柱填料对分离的影响
在-20 kV条件下,分别利用3 μm、1.7 μm及2.7 μm的核壳型Halo颗粒填充的C18柱(均来自苏州环球色谱有限责任公司)对8种单糖衍生物进行分离。结果表明,采用3 μm C18柱时,27 min左右全部出峰,除葡萄糖和半乳糖、阿拉伯糖和木糖两组同分异构体没有完全分离,其余单糖均实现分离;采用新型2.7 μm Halo核壳型填料的C18柱时,柱效提高,峰形改善,所有单糖均达到基本分离,24 min全部出峰;采用1.7 μm C18柱时,各峰分离度进一步提高,所有单糖均达到完全分离,但由于填料粒径小,反压大,保留时间大大延长至75 min左右。综合考虑,选择2.7 μm Halo核壳型填料的C18柱来进行进一步的分离。
2.1.6流动相对分离的影响
选择2.7 μm C18核壳型Halo柱,在-20 kV条件下,分别用20∶80、18∶82和16∶84的乙腈-醋酸铵的流动相对8种单糖衍生物进行分离。结果表明,有机相比例降低,单糖分离度提高,保留时间延长;有机相比例增大加快洗脱,葡萄糖与半乳糖无法分离;综合考虑,选择乙腈-醋酸铵之比为18∶82,此时两组同分异构体分离度为1.3,达到基本分离。
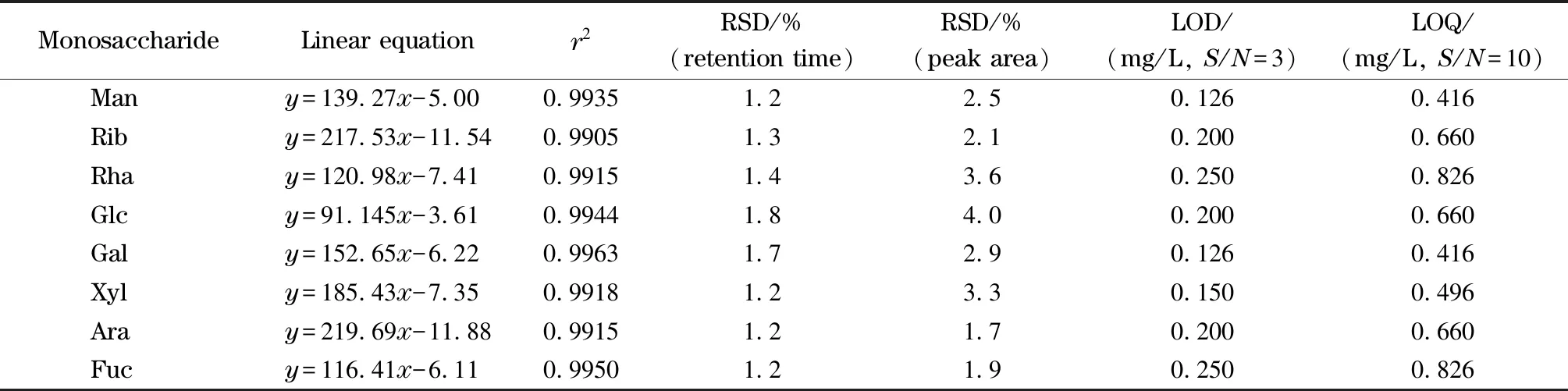
表 2 8种单糖的线性关系、重复性和检出限、定量限分析结果
2.1.7色谱条件的确定
综上所述,经过条件探索与优化,选择Halo-2.7 μm核壳型C18毛细管色谱柱,流动相为乙腈-醋酸铵(18∶82, v/v,其中醋酸铵浓度50 mmol/L, pH 4.1),加压-20 kV,如图1所示,在24 min内实现了8种单糖衍生物的分离。

图 1 8种单糖衍生物混合标准品的pCEC分离色谱图Fig. 1 pCEC chromatogram of a mixture of the eight monosaccharide derivatives Column: C18-Halo (20 cm×100 μm, 2.7 μm); mobile phases: ACN-50 mmol/L pH 4.1 ammonium acetate (18∶82, v/v); flow rate: 0.1 mL/min; split ratio: 1∶400; detection wavelength: 250 nm; applied voltage: -20 kV. Peak identifications: PMP. 1-phenyl-3-methyl-5-pyrazolone; 1. mannose, Man; 2. ribose, Rib; 3. rhamnose, Rha; 4. glucose, Glc; 5. galactose, Gal; 6. xylose, Xyl; 7. arabinose, Ara; 8. fucose, Fuc.
2.2 方法验证
分别将0.30、0.20、0.15、0.12以及0.10 g/L的标准单糖混合液,按照1. 2. 2节方法进行衍生,并按照1.4节色谱条件进行检测,各浓度下8种单糖重复测定3次。将峰面积平均值y对单糖质量浓度x进行线性回归得到线性方程及相关系数(r2);按色谱图中信噪比(S/N)=3,得到检出限(LOD);按S/N=10,得到定量限(LOQ);选择0.30 g/L的标准单糖混合液进样6次,记录8种单糖PMP衍生物的保留时间及峰面积,计算RSD,考察精密度。结果(见表2)表明该方法测定8种单糖具有较好的线性关系和良好的重复性。可用于样品中游离单糖或多糖的单糖组成测定。
2.3 响应面法优化提取工艺
超声辅助提取法因其提取时间短,效率较高,在合适的功率下对提取物破坏小,与传统的浸提方法相比具有较多优势[27,28],因而被广泛应用于有效成分的提取。已有较多研究证明,在超声辅助提取过程中,液料比、超声温度、超声时间及超声功率均对提取率有所影响[20]。
在单因素试验结果的基础上,利用Design-Expert 8.0软件的Box-Behnken设计进行响应面优化实验,通过对实验结果进行多元二次回归拟合,分别建立了粉葛多糖和柴葛多糖的提取工艺参数回归模型。
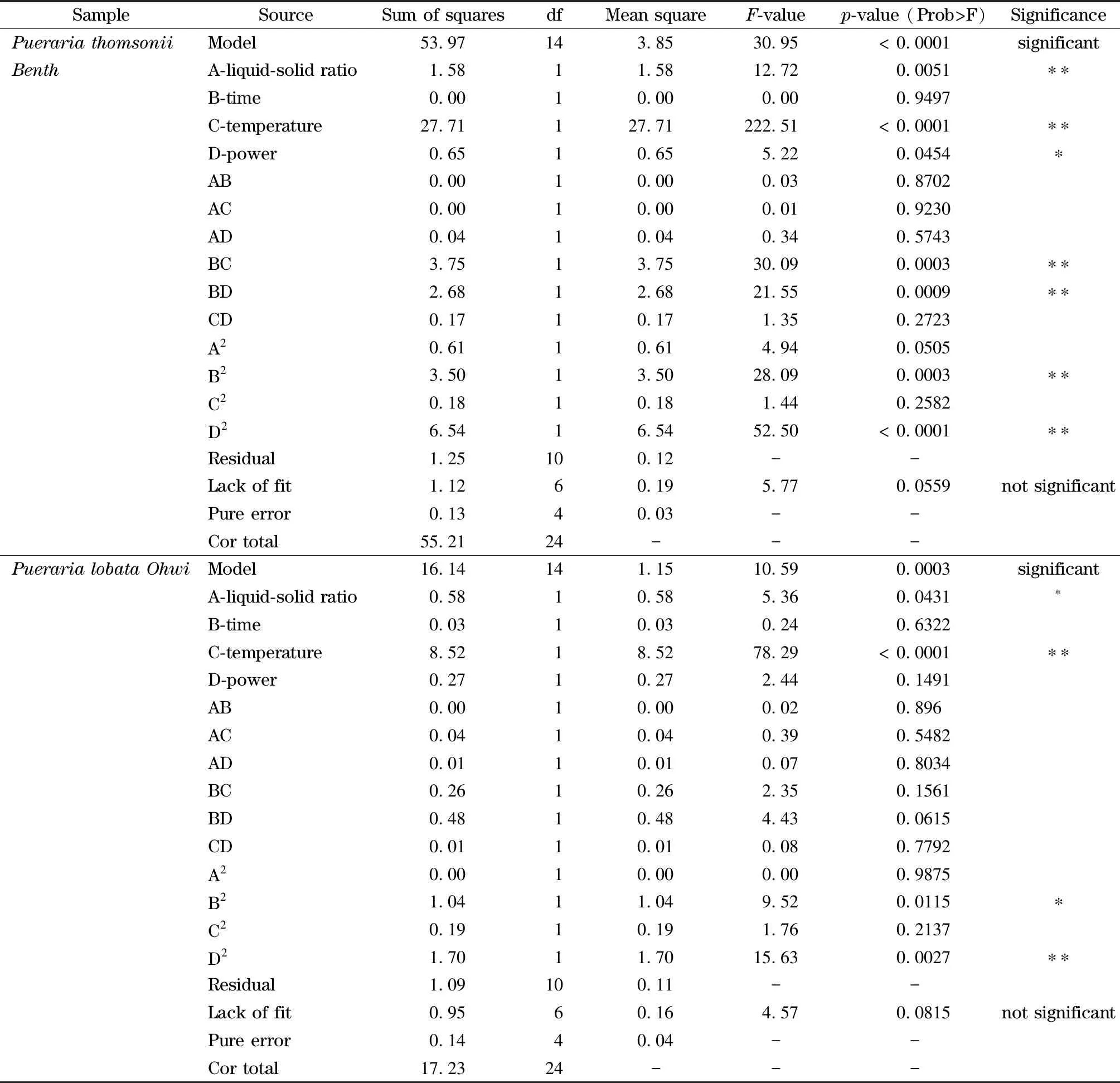
表 3 粉葛多糖和柴葛多糖提取响应面方差分析结果
粉葛多糖的提取率回归方程为:Extraction rate=9.81+0.44A-0.008 314B+1.68C+0.35D-0.028AB+0.017AC-0.15AD-1.25BC-1.18BD-0.20CD-0.35A2-0.82B2-0.19C2-1.27D2;柴葛多糖的提取率回归方程为:Extraction rate=6.94+0.27A+0.059B+0.93C+0.22D-0.021AB+0.10AC+0.062AD-0.33BC-0.50BD-0.047CD-0.002 385A2-0.45B2-0.20C2-0.65D2。

图 2 四因素交互影响粉葛多糖提取率的响应面三维图Fig. 2 Response surface 3D plots of the extraction rate of Pueraria thomsonii a. time (B) and liquid/solid ratio (A); b. temperature (C) and liquid/solid ratio (A); c. power (D) and liquid/solid ratio (A); d. temperature (C) and time (B); e. power (D) and time (B); f. power (D) and temperature (C).

图 3 四因素交互影响柴葛多糖提取率的响应面三维图Fig. 3 Response surface 3D plots of the extraction rate of Pueraria lobata OhwiFor a-f, see Fig. 2.
粉葛多糖和柴葛多糖的超声辅助提取响应面方差分析结果见表3。由方差分析结果可知,两个模型的p值都达到了极显著水平,失拟项均大于0.05,说明失拟项不显著,所建立的回归模型能充分反应实际情况。回归模型的总决定系数(R2)为0.977 4和0.936 8,说明葛根多糖的提取率与模型预测结果有良好的一致性。由F检验值可知,A液料比、B时间、C温度和D功率4项对两种葛根多糖提取率的影响大小顺序为:C>A>D>B,即超声温度对得率影响最大,其次为液料比、超声功率和时间,对粉葛多糖:时间的影响不显著,模型中的交互项BC和BD、二次项B2和D2都达到了极显著水平;对柴葛多糖:功率和时间的影响不显著,模型中仅有二次项B2和D2达到了显著水平。
四因素交互对两种葛根多糖提取率的响应面三维图如图2和图3所示,对4个因素两两交互作用进行分析,交互曲面的陡峭程度及等值线的椭圆程度可以反映因素影响及交互效应的强弱[29]。图2d和图2e中曲面较为陡峭且等值线椭圆程度高,表明时间和温度、时间和功率两组因素交互作用强,与粉葛多糖的方差分析结果一致。图3中等值线椭圆程度比图2中小,表明柴葛多糖提取率受四因素交互影响较粉葛多糖相对不显著。
结合软件预测分析得到的最佳条件及设备实际情况,确定葛根多糖的最佳提取工艺条件为:粉葛多糖液料比20 mL/g,柴葛多糖液料比40 mL/g,超声温度90 ℃,超声时间30 min,超声功率180 W。以此条件对实际样品进行多糖提取。
2.4 葛根实际样品单糖组成的pCEC分析
应用所建立的加压毛细管电色谱法,对2种葛根样品的单糖组成进行了测定,结果见图4。将实际样品谱图与图1对应可知:粉葛(见图4a)主要由葡萄糖、甘露糖、鼠李糖和岩藻糖组成,根据峰面积计算得到4种单糖物质的量之比为0.16 (Man)∶0.14 (Rha)∶1.00 (Glc)∶0.07 (Fuc);柴葛(见图4b)主要由葡萄糖和甘露糖组成,2种单糖物质的量之比为0.70 (Man)∶1.00 (Glc)。实际样品的单糖组成测定结果与赵丹等[17]报道的离子色谱方法测定的葛根样品中岩藻糖、鼠李糖、葡萄糖的结果大致相符,与周礼仕等[9]报道的不同产地的葛根多糖单糖组成结果有所差异,判断可能由于葛根产地不同而导致单糖组成差异。

图 4 葛根多糖水解液衍生物的pCEC分离图Fig. 4 pCEC chromatograms of hydrolyzed derivatives of actual polysaccharide samplesa. Pueraria thomsonii Benth; b. Pueraria lobata Ohwi.
3 结论
本实验通过响应面法对葛根多糖超声提取法中液料比、超声温度、超声时间和超声功率进行了工艺探究,研究了4个因素对葛根多糖提取率的影响,建立了柱前衍生加压毛细管电色谱测定8种中性单糖的新方法,对该法中缓冲盐浓度、缓冲盐pH、施加电压、色谱柱以及流动相比例进行了探索与优化,并对葛根多糖样品的单糖组成进行了分析与测定。本方法简单、快速、分离效果好、柱效高,可作为样品中游离单糖或多糖的单糖组成测定的新方法。
猜你喜欢
云南化工(2020年11期)2021-01-14
源流(2020年6期)2020-08-03
农家之友(2020年2期)2020-05-19
天然产物研究与开发(2019年1期)2019-03-01
中成药(2017年6期)2017-06-13
食品界(2016年3期)2016-09-10
天然产物研究与开发(2016年1期)2016-06-05
杂草学报(2015年2期)2016-01-04
中药与临床(2015年5期)2015-12-17
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
