
新型meso位芘取代氟硼二吡咯衍生物的合成及光敏活性
2020-09-21 11:59杨小松邹遥海梁力曼
合成化学 2020年9期
杨小松, 邹遥海, 梁力曼, 孟 迪, 彭 飞, 牛 奎*
(1. 河北科技师范学院 a. 化学工程学院; b. 分析测试中心,河北 秦皇岛 066004)
单线态氧(1O2)作为一种处于最低激发态的分子氧,具有较高的能量和很强的亲电性,可参与烯烃的加成、Diels-Alder环加成以及杂原子化合物的氧化等反应,且反应具有条件温和、位点选择性强和副反应少的特点[1-3]。此外,1O2有着更为经济和洁净的来源,即在光敏化剂的作用下对基态氧进行辐照即可产生。因此,利用太阳辐射光作为能量来源,以染料敏化过程中产生的1O2作为活性氧物种,能够实现有机氧化反应过程中的低能耗和无重金属排放,符合环境友好性和能源利用可持续性的两大绿色化学理念。
氟硼二吡咯(BODIPY)荧光染料具有高摩尔消光系数、高荧光量子产率、大的斯托克斯位移以及良好的光和化学稳定性,近年来在染料敏化太阳能电池[4]、DNA标记和测序[5]、荧光探针[6]以及癌症的光动力疗法(PDT)[7]等领域的应用备受关注。无取代的BODIPY呈平面结构,在光辐射下不易产生激发三重态,因此其1O2的生成效率极低。在BODIPY母体结构的meso位引入电子给体是获得BODIPY三重态光敏剂的重要方式,组装的电子给-受体复合物中存在光诱导电子转移(PET)或分子内电荷转移(ICT),转移后的电荷复合(CR)过程可以大幅提升染料的三重态生成几率[8-9]。提高BODIPY染料光敏活性的另一典型途径是用卤素原子对BODIPY的分子结构进行修饰,借助重原子效应促进染料分子由激发单重态至三重态的系间窜越(ISC),进而增大BODIPY的1O2量子产率。已有相关研究[10-11]表明,将碘或溴原子引入BODIPY母环可有效提高染料分子的ISC效率,且碘原子取代的BODIPY表现出更加显著的1O2介导的光敏催化效能。此外,将多个BODIPY母环通过共价键进行桥连,可以利用联BODIPY分子受激发时产生的PET和CR过程对染料的三重态产生增强效应[12]。对meso-βBODIPY二聚体的相关研究表明,子单体结构的正交性和溶剂极性是影响其三重态产生效率和1O2生成能力的重要因素[13-14]。
制备了芘-BODIPY的电子给-受体结构,并在此基础上进行了BODIPY的二聚体化和重原子化,以期提升染料的三重态产生效率,得到具有高光敏氧化活性的新型BODIPY光敏剂。由于在meso位引入发色团几乎不会影响BODIPY的电子吸收光谱[15],因此组装芘-BODIPY双发色团体系可有效地吸收利用350~450 nm波段的能量,这对于在太阳光诱导的光氧化反应中光敏氧化效率的提升具有积极作用和现实意义。
合成路线如Scheme 1所示,以1-芘甲醛为起始原料分别与2,4-二甲基吡咯和吡咯依次经过缩合反应、氧化脱氢、氟硼配位后,得到meso位连接富电子芘环的BODIPY染料1和2。1经Vilsmeier-Haack反应在β位引入醛基生成中间体3,再分别与2,4-二甲基吡咯和吡咯反应得到两种新型的BODIPY二聚体1a和1b。2以相同路径经由中间体4得到新型的BODIPY二聚体2a和2b。将1通过碘化反应在BODIPY的β位引入碘原子,生成新型的单碘代衍生物1c和双碘代衍生物1d。各产物的结构经1H NMR、13C NMR和HR-MS(APCI)表征。测试了各染料的光物理性质并以1,3-二苯基异苯并呋喃(DPBF)为1O2捕获剂初步考察了产物的光敏氧化性能。
1 实验部分
1.1 仪器与试剂
XT5B型显微熔点测定仪;F97型荧光分光光度计;UV-5500型紫外可见分光光度计;Bruker AVANCE III 600 MHz型核磁共振波谱仪(CDCl3为溶剂,TMS为内标);LTQ Orbitrap XL组合型离子阱和轨道阱质谱仪。
1-芘甲醛、2,3-二氯-5,6-二氰对苯醌购自梯希爱,其余所用试剂均为分析纯或化学纯。
1.2 合成
(1)1和2的合成(以1为例)
室温氮气保护下,称取1-芘甲醛2.53 g(11.0 mmol) 和2,4-二甲基吡咯2.0 g(21.1 mmol)溶解于500 mL二氯甲烷中,再滴加三氟乙酸(200 μL)与二氯甲烷(6.20 mL)的混合溶液。搅拌3 h后逐滴加入含有2,3-二氯-5,6-二氰对苯醌2.5 g(11 mmol)的二氯甲烷(100 mL)。滴毕,搅拌20 min后加入三乙胺(15 mL),随后滴加三氟化硼的乙醚(15 mL),滴毕继续反应3 h(TLC检测)。反应液以去离子水(3×500 mL)和饱和碳酸氢钠溶液(2×100 mL)洗涤,有机相以无水硫酸镁干燥,过滤后蒸除溶剂。残余物经硅胶柱层析[洗脱剂:V(二氯甲烷)/V(正己烷)=1/1]纯化得1。
将上述步骤中的2,4-二甲基吡咯替换为等量吡咯,用类似方法合成2。
1: 橙黄色固体1035 mg,收率21%,m.p.296~297 ℃;1H NMR(CDCl3, 600 MHz)δ: 8.29(d,J=7.8 Hz, 1H), 8.26(d,J=7.6 Hz, 1H), 8.21(d,J=7.5 Hz, 1H), 8.17(d,J=8.9 Hz, 1H), 8.14(d,J=8.9 Hz, 1H), 8.07(d,J=7.6 Hz, 1H), 8.05(s, 1H), 8.03(d,J=9.1 Hz, 1H), 7.90(d,J=7.8 Hz, 1H), 5.95(s, 2H), 2.63(s, 6H), 0.89(s, 6H);13C NMR(CDCl3, 151 MHz)δ: 155.72, 143.23, 140.55, 132.27, 131.73, 131.27, 131.02, 129.34, 128.92(2C), 128.33(2C), 127.29, 126.47, 125.82(2C), 125.77(2C), 125.69, 125.33(2C), 124.68, 124.52, 124.12, 121.2, 14.69(2C), 13.87(2C); HR-MS(APCI)m/z: Calcd for C29H23N2BF{[M-F]+}429.1938, found 429.1938。
2: 橙红色固体733 mg,收率17%,m.p.271~272 ℃;1H NMR(CDCl3, 600 MHz)δ: 8.28(d,J=7.6 Hz, 1H), 8.26(d,J=7.8 Hz, 1H), 8.22(s, 1H), 8.20(s, 1H), 8.16(s, 1H), 8.08(s, 1H), 8.07(s, 1H), 8.04(s, 1H), 8.03(s, 1H), 8.01(s, 2H), 6.62(d,J=3.5 Hz, 2H), 6.46(d,J=2.9 Hz, 2H);13C NMR(CDCl3, 151 MHz)δ: 144.43(2C), 132.52, 131.75(2C), 129.00(2C), 128.52(2C), 127.84(2C), 127.16(2C), 126.65(2C), 126.17(2C), 125.95(2C), 124.98(2C), 124.00(2C), 118.81(2C); HR-MS(APCI)m/z: Calcd for C25H15N2BF{[M-F]+} 373.1312, found 373.1277。
(2)3和4的合成(以3为例)
冰浴氮气保护下,三氯氧磷(7.5 mL)缓慢滴加至N,N-二甲基甲酰胺(7.5 mL)中,搅拌5 min后缓慢升至室温反应20 min。将溶有1269 mg (0.6 mmol)的1,2-二氯乙烷(70 mL)缓慢滴加至体系。滴毕,温度升至50 ℃反应6 h(TLC检测)。冷却至室温,反应液以冷的饱和碳酸氢钠溶液(2×200 mL)和去离子水(2×200 mL)洗涤,有机相以无水硫酸钠干燥,过滤后蒸除溶剂。粗产物经硅胶柱层析[洗脱剂:V(二氯甲烷)/V(正己烷)=2/1]纯化得中间体3。
将上述步骤中的1替换为等量2,用类似方法合成4。
3: 紫红色固体180 mg,收率63%,m. p.>300 ℃;1H NMR(CDCl3, 600 MHz)δ: 9.94(s, 1H), 8.33(d,J=7.8 Hz, 1H), 8.29(d,J=7.6 Hz, 1H), 8.24(d,J=7.5 Hz, 1H), 8.21(d,J=8.9 Hz, 1H), 8.16(d,J=8.9 Hz, 1H), 8.09(d,J=7.4 Hz, 1H), 8.07(s, 1H), 7.95(d,J=9.1 Hz, 1H), 7.88(d,J=7.8 Hz, 1H), 6.12(s, 1H), 2.90(s, 3H), 2.69(s, 3H), 1.19(s, 3H), 0.92(s, 3H);13C NMR(CDCl3, 151 MHz)δ: 185.85, 161.84, 156.70, 147.28, 143.07, 142.65, 134.94, 132.11, 131.28, 130.94, 130.50, 129.42, 129.19, 128.74, 128.24, 127.24, 126.70, 126.39, 126.12, 125.97, 125.51, 125.46, 124.73, 124.46, 124.06, 123.55, 15.20, 14.36, 13.21, 10.98; HR-MS(APCI)m/z: Calcd for C30H24N2OBF2{[M+H]+}477.1950, found 477.1927。
4: 紫红色固体202 mg,收率80%,m. p.>300 ℃;1H NMR(CDCl3, 600 MHz)δ: 9.73(s, 1H), 8.30(d,J=7.8 Hz, 1H), 8.28(d,J=7.7 Hz, 1H), 8.22(d,J=7.5 Hz, 1H), 8.19(d,J=9.0 Hz, 1H), 8.15(t,J=7.2 Hz, 1H), 8.09(s, 1H), 8.06(s, 1H), 7.98(d,J=9.1 Hz, 1H), 7.86(d,J=7.8 Hz, 1H), 6.74(s, 1H), 6.62(s, 1H), 6.53(s, 1H), 6.46(s, 1H), 6.01(s, 1H);13C NMR(CDCl3, 151 MHz)δ: 177.69, 145.46, 143.38, 136.41, 135.47, 134.91, 132.11, 130.84, 129.36, 128.95, 128.65, 127.95, 127.47, 126.77, 126.11, 125.60, 125.13, 124.91, 124.73, 124.46, 124.06, 123.92, 123.53, 123.20, 122.95, 121.72; HR-MS(APCI)m/z: Calcd for C26H16N2OBF2{[M+H]+}421.1324, found 421.1301。
(3)1a,1b,2a,2b的合成(以1a为例)
氮气保护下,将2,4-二甲基吡咯180 mg(1.90 mmol)、三氟乙酸(9 μL)加入溶有386 mg(0.18 mmol) 的二氯甲烷(10 mL)中,反应4 h。用0.2 mol/L 氢氧化钠溶液(30 mL)淬灭反应后,加入二氯甲烷(50 mL)萃取,有机相以无水硫酸镁干燥、过滤。向溶液中加入2,3-二氯-5,6-二氰对苯醌60 mg(0.24 mmol),反应1 h后转入冰浴,加入三乙胺(1 mL)和三氟化硼乙醚(1 mL),反应20 min后再升至室温反应3 h(TLC检测)。反应液以去离子水(3×150 mL)洗涤,有机相以无水硫酸镁干燥,过滤后蒸除溶剂。粗产物经硅胶柱层析[洗脱剂:V(二氯甲烷)/V(正己烷)=1/1]纯化得1a。
将步骤中的2,4-二甲基吡咯替换为等量吡咯,用类似方法合成1b。将3替换为等量4,用类似方法分别合成2a和2b。
1a: 紫红色固体24 mg,收率19%,m.p.>300 ℃;1H NMR(CDCl3, 600 MHz)δ: 8.30(d,J=7.8 Hz, 1H), 8.26(d,J=7.5 Hz, 1H), 8.22(d,J=7.5 Hz, 1H), 8.17(d,J=8.9 Hz, 1H), 8.13(d,J=8.9 Hz, 1H), 8.07(d,J=7.3 Hz, 1H), 8.05(s, 1H), 7.99(d,J=9.1 Hz, 1H), 7.92(d,J=7.8 Hz, 1H), 6.05(s, 1H), 5.98(s, 1H), 5.92(s, 1H), 2.69(s, 3H), 2.51(s, 3H), 2.48(s, 3H), 1.78(s, 3H), 1.73(s, 3H), 1.28(s, 3H), 0.95(s, 3H), 0.71(s, 3H);13C NMR(CDCl3, 151 MHz)δ: 157.90, 154.61, 149.70, 144.56, 141.35, 140.31, 137.46, 132.47, 132.28, 130.94, 130.70, 130.23, 129.85, 128.10(2C), 127.57(2C), 126.21(2C), 125.59(2C), 125.03(2C), 124.80, 124.66(2C), 124.43(2C), 123.74, 123.42, 122.41, 121.52, 120.16(2C), 13.86, 13.66, 13.55, 13.11, 12.98, 12.86, 11.76, 10.50; HR-MS(APCI)m/z: Calcd for C42H36N4B2F3{[M-F]+}675.3078, found 675.3026。
1b: 深紫色固体17 mg,收率15%,m.p.>300 ℃;1H NMR(CDCl3, 600 MHz)δ: 8.32(s, 1H), 8.27(s, 1H), 8.21(s, 1H), 8.17(d,J=13.0 Hz, 1H), 8.13(s, 1H), 8.07(s, 1H), 7.94(s, 1H), 7.83(s, 1H), 7.79(s, 1H), 7.54(s, 1H), 7.26(s, 1H), 6.88(s, 1H), 6.76(s, 1H), 6.48(s, 1H), 6.40(s, 1H), 6.09(s, 1H), 2.70(s, 3H), 2.56(s, 3H), 0.95(s, 3H), 0.88(s, 3H);13C NMR(CDCl3, 151 MHz)δ: 160.19, 152.05, 146.36, 144.11, 141.77, 139.89, 135.49, 134.13, 132.05, 131.54, 131.30, 130.94(2C), 130.84(2C), 129.38(2C), 129.20, 128.68(2C), 128.51, 127.26(2C), 126.69, 126.13, 125.91, 125.55(2C), 124.78, 124.50, 123.56, 123.30, 118.48(2C), 15.09, 14.26, 13.73, 12.79; HR-MS(APCI)m/z: Calcd for C38H28N4B2F3{[M-F]+}619.2452, found 619.2404。
2a: 深紫色固体18 mg,收率16%,m.p.>300 ℃;1H NMR(CDCl3, 600 MHz)δ: 8.29(d,J=7.6 Hz, 1H), 8.27(d,J=7.9 Hz, 1H), 8.23(d,J=7.6 Hz, 1H), 8.21(d,J=9.0 Hz, 1H), 8.15(d,J=4.3 Hz, 1H), 8.08(d,J=3.3 Hz, 1H), 8.07(s, 1H), 8.05(d,J=2.3 Hz, 1H), 8.04(d,J=3.7 Hz, 1H), 7.84(s, 1H), 7.72(dd,J=5.7 Hz, 3.3 Hz, 1H), 7.53(dd,J=5.7 Hz, 3.3 Hz, 1H), 6.81(d,J=4.2 Hz, 1H), 6.59(t,J=4.6 Hz, 1H), 6.53(s, 1H), 5.95(d,J=7.4 Hz, 1H), 1.82(s, 3H), 1.78(s, 3H), 1.57(s, 6H);13C NMR(CDCl3, 151 MHz)δ: 167.74, 155.79, 147.19, 141.23, 133.53, 133.28, 132.82, 132.33, 131.28, 130.93(2C), 130.63(2C), 130.41, 129.62, 129.30, 128.86(2C), 128.80, 127.60(2C), 127.17, 127.08, 126.78(2C), 126.48, 126.15, 124.63(2C), 124.53, 124.18, 124.13, 121.45, 120.20, 15.41, 15.25, 13.74, 12.69; HR-MS(APCI)m/z: Calcd for C38H28N4B2F3{[M-F]+}619.2452, found 619.2457。
2b: 深紫色固体13 mg,收率12%,m.p.>300 ℃;1H NMR(CDCl3, 600 MHz)δ: 8.27(s, 2H), 8.22(s, 2H), 8.16(s, 1H), 8.08(s, 1H), 8.04(s, 1H), 8.01(s, 2H), 7.70(s, 1H), 7.18(s, 1H), 7.11(s, 1H), 7.03(s, 1H), 6.85(s, 1H), 6.73(s, 1H), 6.64(s, 2H), 6.47(s, 2H), 5.74(s, 1H);13C NMR(CDCl3, 151 MHz)δ: 146.55, 146.44, 136.52, 132.53, 131.75(2C), 131.29, 130.83, 130.79, 130.75, 130.41, 129.00(2C), 128.53(2C), 127.85, 127.77, 127.71, 127.16(2C), 126.65(2C), 126.18(2C), 125.96(2C), 124.98(2C), 124.57, 124.29, 124.21, 124.01(2C), 118.76; HR-MS(APCI)m/z: Calcd for C34H20N4B2F3{[M-F]+}563.1826, found 568.1841。
(4)1c和1d的合成(以1c为例)
称取碘203 mg(0.8 mmol)和碘酸141 mg(0.8 mmol)溶解于水(1 mL)中,加入溶有1448 mg(1.0 mmol)的乙醇(100 mL),升温至60 ℃反应20 min。将反应液倾入饱和硫代硫酸钠水溶液(100 mL),以氯仿(2×100 mL)萃取,合并有机相,无水硫酸钠干燥,减压蒸除溶剂。粗产物经硅胶柱层析[洗脱剂:V(氯仿)/V(正己烷)=1/1]纯化得1c。
将步骤中碘和碘酸的用量均变为2.5 mmol,用类似方法合成1d。
1c: 深紫色固体431 mg,收率75%, m.p.>300 ℃;1H NMR(CDCl3, 600 MHz)δ: 8.30(d,J=7.8 Hz, 1H), 8.28(d,J=7.7 Hz, 1H), 8.22(d,J=7.5 Hz, 1H), 8.19(d,J=9.0 Hz, 1H), 8.15(d,J=8.9 Hz, 1H), 8.08(d,J=7.6 Hz, 1H), 8.07(m, 1H), 7.98(d,J=9.1 Hz, 1H), 7.86(d,J=7.8 Hz, 1H), 6.01(s, 1H), 2.71(s, 3H), 2.64(s, 3H), 0.91(s, 3H), 0.88(s, 3H);13C NMR(CDCl3, 151 MHz)δ: 158.03, 154.79, 145.21, 143.36, 140.48, 132.73, 131.90, 131.68, 131.26, 130.98, 129.26, 129.16, 129.02, 128.52, 127.26(2C), 126.58, 125.92, 125.82, 125.66, 125.40, 124.69, 124.48, 123.87, 122.36, 16.09, 15.90, 14.88, 14.11; HR-MS(APCI)m/z: Calcd for C29H22N2BFI{[M-F]+}555.0905, found 555.0900。
1d: 深紫色固体672 mg,产率96%,m.p.>300 ℃;1H NMR(CDCl3, 600 MHz)δ: 8.30(t,J=7.7 Hz, 2H), 8.22(dd,J=12.0 Hz, 8.3 Hz, 2H), 8.16(d,J=8.9 Hz, 1H), 8.09(d,J=7.6 Hz, 1H), 8.06(d,J=9.3 Hz, 1H), 7.94(d,J=9.1 Hz, 1H), 7.83(d,J=7.7 Hz, 1H), 2.73(s, 6H), 0.90(s, 6H);13C NMR(CDCl3, 151 MHz)δ: 156.98(2C), 145.40, 140.46, 132.06(2C), 131.25, 130.93, 129.38(2C), 129.19, 128.73(2C), 127.22(2C), 126.67(2C), 126.06, 125.93(2C), 125.47(2C), 124.69, 124.43, 123.64, 16.40(2C), 16.14(2C); HR-MS(APCI)m/z: Calcd for C29H21N2BFI2{[M-F]+}680.9871, found 680.9845。
1.3 光学性质
(1) 光物理参数
分别取少量各样品于二氯甲烷中超声充分溶解。取3 mL溶液加入石英比色皿,调整溶液浓度使最大吸收波长(λabs)处的吸光度<0.5,测量250~700 nm波长范围内的紫外-可见吸收光谱并记录λabs。调整溶液浓度控制激发波长在475 nm处的吸光度为0.05~0.1,取3 mL溶液加入石英四通光比色皿测试激发光谱和荧光发射光谱,记录最大激发波长(λex)和最大发射波长(λem)。
(2) 光敏活性
以二氯甲烷为溶剂,将配置好的DPBF溶液与样品溶液在石英比色皿中混合。调整混合比例,使混合液的DPBF浓度约为30mol/L,同时在样品对应最大吸收波长处(490~510 nm)吸光度为1.0左右。以白炽灯为模拟光源,固定光源与比色皿距离为20 cm,记录不同照射时间后溶液的吸收光谱,根据410 nm处吸光度和照射时间绘制光敏反应动力学曲线。
2 结果与讨论
2.1 BODIPY衍生物的光物理性质
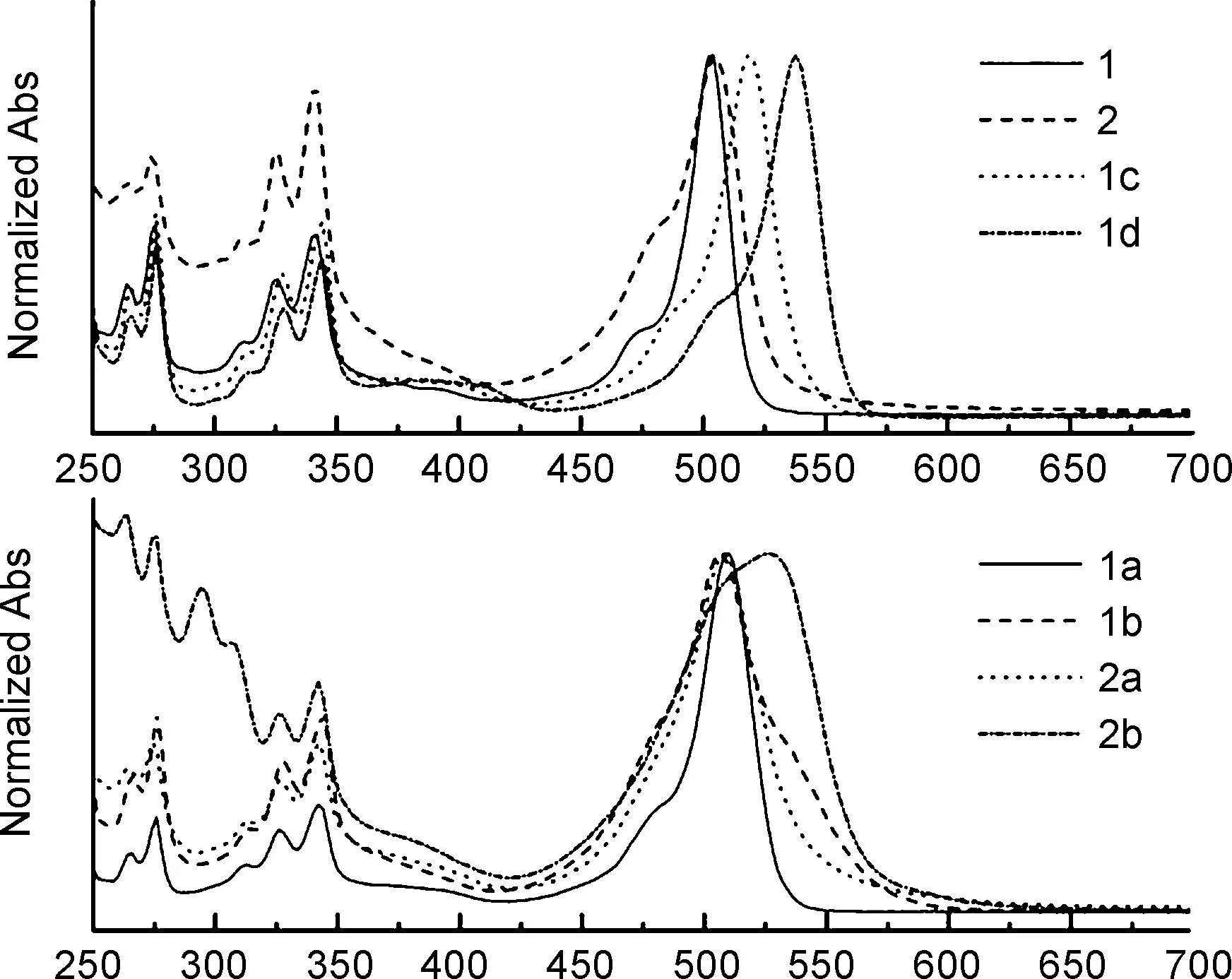
图1为所合成BODIPY衍生物在二氯甲烷溶液中的归一化吸收光谱,各产物在250~350 nm的吸收峰组与芘发色团的特征吸收峰相吻合。与母体1相比,两种碘代产物1c和1d的最大吸收波长有着显著的红移,但BODIPY发色团吸收峰的峰形未有明显改变。四种二聚体产物的最大吸收波长与对应母体相比也有着不同程度的红移,且除1a外其它产物的吸收峰半峰宽明显变大,表明1b,2a和2b中的BODIPY二聚体结构存在着更强的π-体系间的激子偶合作用[16]。
/nm
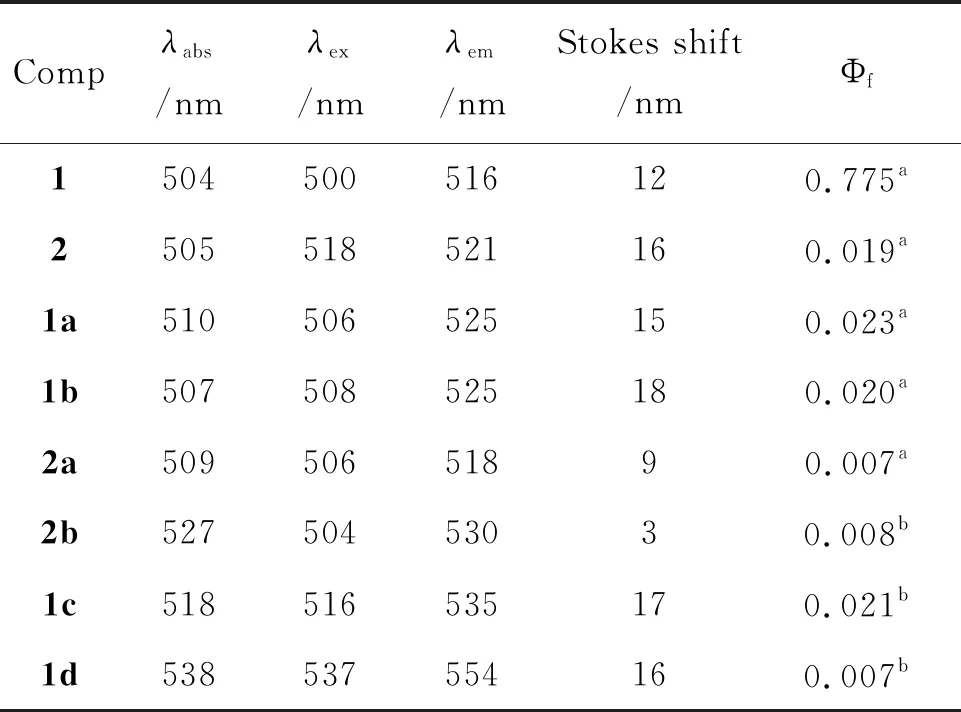
表1 BODIPY衍生物在二氯甲烷溶液中的光物理参数
/nm
Time/s
表1给出了所合成BODIPY衍生物在二氯甲烷溶液中的光物理参数。产物的最大发射波长在516~554 nm,斯托克斯位移均小于20 nm。1的荧光量子产率高达0.775,远高于2的0.019,这是由于1中BODIPY母环1,7位的甲基可以有效限制meso-位芘环的自由转动,减少了分子非辐射失活的途径[17]。
2.2 BODIPY衍生物的光敏活性
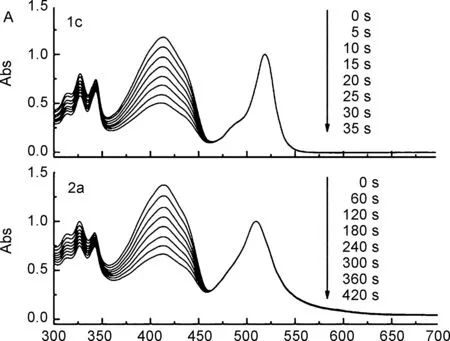
DPBF与1c/2a在二氯甲烷中吸收光谱随光照时间的变化如图2A所示。DPBF的最大吸收峰位于410 nm处,随光照时间增加该处的吸光度迅速降低,同时BODIPY自身吸收峰强度不变。将溶液在暗处下进行对比实验,其吸收光谱不随时间改变,证实在光照下1c和2a均能有效敏化产生1O2,从而实现对DPBF的光敏氧化降解。
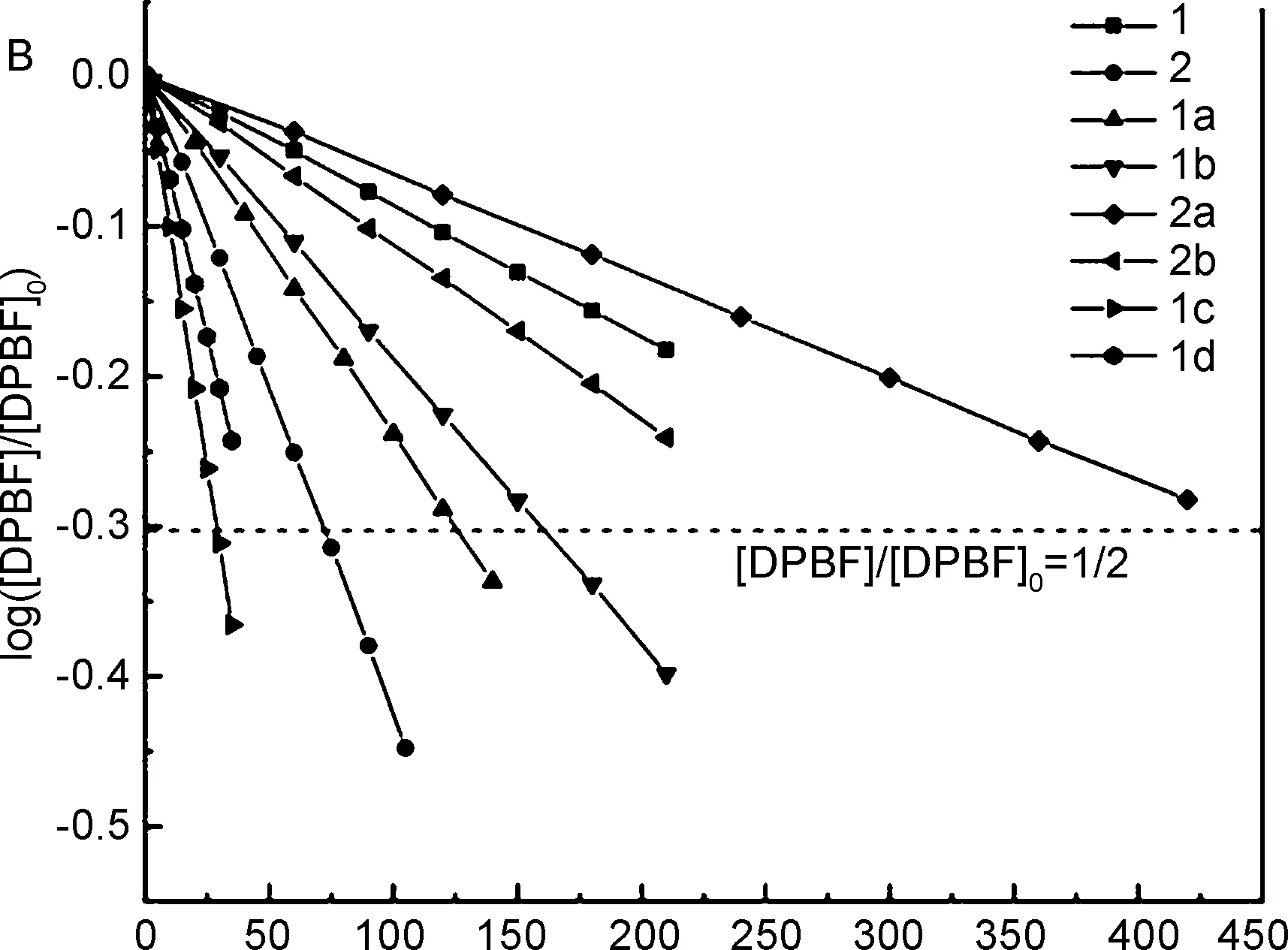
由图 2B可知,所有衍生物的反应动力学曲线都呈现良好的线性关系,DPBF的光敏氧化降解具有零级反应的动力学特点。由拟合直线的斜率大小得到各产物光敏氧化活性的顺序为:1c>1d>2>1a>1b>2b>1>2a。
合成了两种含有芘-BODIPY双发色团结构的染料1和2,通过对BODIPY母核进行二聚体化及重原子化设计合成了6种新型BODIPY衍生物(1a~1d,2a,2b)。所有碘代产物和二聚体含有芘的特征吸收,对应BODIPY发色团的吸收峰相较母体红移,荧光量子产率相较母体降低。光敏氧化降解实验表明:1的重原子化和二聚体化均能有效提升染料的光敏氧化性能,其中光敏活性最强的是单碘代产物1c,对DPBF的光敏降解速率是母体1的12.0倍,半数降解时间仅为29 s;2对DPBF的光敏降解速率是1的4.9倍,光照72 s即可实现半数降解。
猜你喜欢
中国饲料(2022年5期)2022-04-26
化学工程师(2022年3期)2022-04-19
化学工业与工程(2022年1期)2022-03-29
湖南科技学院学报(2021年3期)2021-10-21
建材发展导向(2021年13期)2021-07-28
炎黄地理(2021年12期)2021-01-04
化工管理(2020年8期)2020-06-02
生物工程学报(2020年1期)2020-03-12
大众科学(2017年10期)2017-12-18
科技与创新(2015年20期)2015-10-29
