
利用示踪粒子及膨胀流变学研究β-乳球蛋白聚集体的界面行为
2020-09-21 08:15张岳梅高志明NISHINARIKatsuyoshi方亚鹏
食品科学 2020年17期
叶 晶,李 静,张岳梅,黄 萍,王 倩,高志明,2,杨 楠,2,*,NISHINARI Katsuyoshi,2,方亚鹏,2
(1.湖北工业大学生物工程与食品科学学院,菲利普斯胶体研究中心,湖北 武汉 430068;2.湖北工业大学,湖北省食品胶体国际合作基地,湖北 武汉 430068)
蛋白质是一类重要的生物大分子,具有两亲性,因此可以作为乳化剂稳定油-水界面。蛋白质分子具有多级结构,当对蛋白质进行热加工时,稳定其高级结构的氢键、离子键、疏水相互作用、范德华力以及肽链内的二硫键等分子内相互作用会发生改变,蛋白质高级结构破坏,蛋白质分子内疏水基团暴露,蛋白分子间发生聚集,形成聚集体[1-2]。近年来,关于蛋白质聚集体形成条件和形成机制受到人们的关注。如Schmitt等在pH 7条件下85 ℃加热15 min制备了乳清分离蛋白的蠕虫状聚集体,而在pH 6条件下制得更为致密的球状聚集体[3]。Akkermans等在pH 2条件下,90 ℃加热β-乳球蛋白(β-lactoglobulin,β-lg)5 h后得到纤维状聚集体[4]。
由于尺寸效应及表面结构的差异,蛋白聚集体的界面活性、黏弹性、乳化特性等可能优于天然蛋白质。如Liu Fu等研究由大豆分离蛋白纳米颗粒稳定的乳液表现出良好的抗聚结和稳定性[5]。Gao Zhimin等研究发现β-lg纤维聚集体增强了界面上蛋白分子间的相互作用,能有效提高界面活性[6]。另一方面,蛋白聚集体作为乳化剂在油-水界面形成的蛋白膜结构和流变学特性会影响脂肪消化特性。蛋白膜的交联程度和黏弹性会影响小分子乳化剂如胆盐对界面蛋白的取代,即当油-水界面形成了强烈交联蛋白质膜时,小分子可能很难对其发生取代,从而影响脂肪的消化[7]。Patino等总结了关于β-酪蛋白、酪蛋白酸盐和β-lg在气-水界面的吸附行为和被单甘酯的取代行为,发现不同的蛋白质显示出不同的界面结构,从而影响消化结果[8]。但是目前对蛋白聚集体对脂肪消化的影响研究鲜有报道。
对于蛋白膜结构和流变学特性的研究方法主要包括原子力显微镜、界面剪切流变学、界面膨胀流变学等。如Morris课题组对小分子表面活性剂取代界面处蛋白进行了大量的研究,利用原子力显微镜研究β-酪蛋白和β-lg界面在气-水界面被吐温20取代[9],十二烷基硫酸钠对β-lg取代[10],以及通过模拟生理环境,研究胆盐对界面处β-lg的取代过程和结构变化[11]。Wang Lijun等利用Du Noüy环流变设备研究发现蚕丝蛋白在气-水界面形成的蛋白膜比油-水界面更脆弱,且蛋白质的界面模量可能受到溶剂极性的强烈影响[12]。Cicuta等利用相互成直角的Wilhelmy板测定β-lg和β-酪蛋白在空气-水界面处的表面压力和总黏弹性[13]。Seta等利用界面张力仪测定β-lg在不同界面的膨胀流变学信息,发现β-lg在气-水界面的黏弹性大于在向日葵油-水界面的黏弹性[14]。但由于界面蛋白吸附是一个动态的过程,形成的蛋白膜结构较弱且不均一,上述传统研究方法或很难实现原位测量,或难以给出局部信息。而微流变学作为一种新兴的技术,可实现原位测量,并具有高敏感度[15]。Lee等利用粒子追踪微流变学技术研究了β-lg在油-水及气-水界面上的吸附及成膜过程,给出了这一过程更详细的信息[16-17]。Jordens等利用粒子追踪手段研究了具有较高长径比的β-lg纤维在低pH值(pH 2、3)下的界面性质,揭示了纤维状聚集体形成高弹性界面的特征[18]。但到目前为止,利用界面微流变学系统研究不同蛋白聚集体界面行为的工作较少,并且聚集体对消化影响的研究更鲜见。本实验主要利用示踪粒子微流变学,结合膨胀流变学方法,研究不同质量分数的β-lg及其聚集体在中性pH值条件下的油-水界面吸附和被胆盐小分子替代的行为,旨在通过对比纳米颗粒聚集体(β-lg NP)和纤维状聚集体(β-lg F)的界面吸附和替代过程,揭示蛋白聚集体的界面活性和抵抗胆盐取代能力,进而阐明蛋白质聚集体对乳化和乳液脂肪消化可能产生的影响。
1 材料与方法
1.1 材料与试剂
β-lg(纯度90%、分子质量为18.4 kDa) 美国Davis公司;癸烷(纯度99%) 上海麦克林公司;胆盐(纯度90%以上) 国药集团化学试剂有限公司;直径为(1.0±0.2)μm的荧光微球 美国赛默飞公司;NOA61型紫外固化胶 美国Thorlabs公司。
1.2 仪器与设备
JEM-2100型透射电子显微镜(transmission electron microscope,TEM) 日本电子株式会社;IX72型荧光显微镜 日本奥林巴斯公司;Teclis Tracker型界面流变仪 法国Lyon公司;907 Titrino型全自动电位滴定仪瑞士万通有限公司。
1.3 方法
1.3.1 蛋白聚集体的制备
将β-lg粉末溶解于超纯水中分别配制质量分数为1%和2%的蛋白溶液,在4 ℃条件下过夜溶解,待用。在pH 5.8下,将1%的β-lg溶液于85 ℃加热15 min后,冰浴冷却[19]。冷却后经50 kDa透析袋透析72 h,得到蛋白溶液进行冻干,制得β-lg NP。在pH 2条件下,将2%的β-lg溶液,经搅拌后于80 ℃加热16 h后,冰浴冷却[20]。冷却后通过100 kDa透析袋透析72 h后冻干,制得β-lg F。低pH值制备纤维聚集体过程中,通常认为蛋白先解聚形成分子质量更小的多肽,多肽再进一步聚集形成纤维体。利用透析方法可以将在此过程中产生的未发生转化的肽段去除。
以下实验中,如未指明,则β-lg及其冻干后聚集体样品均用pH 7的磷酸盐缓冲液(phosphate buffered saline,PBS)进行分散溶解。
1.3.2 透射电子显微镜表征
分别将β-lg粉末和制备的β-lg NP、β-lg F溶液及冻干后聚集体样品,溶解于pH 7的PBS中至质量分数为0.02%。利用TEM观察其形貌。β-lg F的长径比则由Fiber Application程序统计TEM图所得[21]。
1.3.3 粒子追踪微流变学分析
蛋白吸附阶段实验中所用样品槽参照文献[16]制作,其具有相同内径(12 mm)的特氟龙环(高度为2.5 mm)嵌于金属铝环上部(高度为4 mm),以在高度为1.5 mm处形成一个平稳的油-水界面。金属环底部与载玻片黏合密封。在实验过程中,上部用盖玻片和绝缘硅脂密封,防止样品挥发。
将直径为1 μm的荧光聚苯乙烯微球分散在体积比为7∶3的水-异丙醇溶液中,体积分数约为0.005%。荧光微球的激发波长542 nm、发射波长612 nm。β-lg、β-lg NP和β-lg F溶液由pH 7的PBS配制,质量浓度分别为25、110、220 μg/mL。
在蛋白质吸附实验中:将适量蛋白溶液注入环内,使液面在特氟龙环与金属环交接处;再向界面上缓慢添加6 μL含有荧光粒子的分散液;最后再向界面上添加约200 μL的癸烷。在环上盖上盖玻片,并用绝缘硅脂进行密封。利用IX72荧光显微镜对界面上荧光粒子的运动进行观察。在40 倍物镜下,控制视野中有50~200 个荧光微粒,保证数据的统计性且示踪粒子之间没有相互作用[22]。
在胆盐取代界面蛋白实验中:为添加胆盐,对样品槽进行改进,在内环外加一个金属铝环,并在内环底部垫上玻璃隔片,形成液体通道,使内环外环体相液体联通。当蛋白质完成吸附后,胆盐溶液注射至外环底部,再通过通道扩散至界面,与蛋白膜发生相互作用。由于较高胆盐质量浓度会引起界面处较大波动,对微流变数据分析产生较大影响,所以胆盐终质量浓度为0.1 mg/mL。另外,低质量浓度胆盐可以减慢胆盐取代界面蛋白的过程,利于观察。
数据采集:槽内样品界面稳定后,荧光粒子的运动由CMOS高速相机采集记录,拍摄帧速率为15 张/s,拍摄时长为30 s,实验温度为(25±2)℃。被记录下来的图像用Matlab程序进行处理及数据分析[23]。通过分析图片中的光学中心确定荧光粒子位置,对比多张连续图片,从而得到界面上示踪粒子随时间的二维运动轨迹及均方位移(mean squared displacement,MSD)(用Δr2(τ)表示,单位为μm2),具体算法参见式(1)[24]。
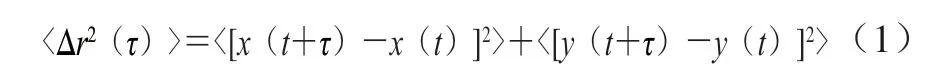
式中:x、y分别为示踪粒子在不同时间图片中的位置;t为实际时间/s;τ为间隔时间/s。示踪粒子的整体漂移通过计算粒子的平均速率消去。<>表示对所有粒子的迟滞时间平均值。本实验中0.06 s<τ<30 s。本实验测定了固定不动粒子的MSD以得到仪器的误差,约为0.001 μm2。
1.3.4 界面流变学测定
本实验的界面流变学测定选用悬滴法,利用界面流变仪分别测定了癸烷-水界面上蛋白吸附过程中界面压π变化(π=γ0-γ,γ0和γ分别为无蛋白和有蛋白吸附时油-水界面张力)以及膨胀模量E(E=Δγ/(ΔA/A0),Δγ为悬滴界面张力变化,A0为悬滴初始表面积,ΔA为悬滴表面积变化)。本实验中体相为蛋白或蛋白聚集体溶液,油相为癸烷。实验采用振荡模式,实验中控制油滴体积为14 μL,控制油滴表面积,振幅变化(ΔA/A0)为10%,振荡频率为0.05 Hz。实验温度为(25±2)℃。
1.3.5 乳液脂肪体外消化模型
利用Tris-HCl缓冲溶液配制质量分数为2%的蛋白溶液。取一定量蛋白溶液,添加中链甘油三酯,使蛋白质量分数为0.5%,中链甘油三酯终质量分数分别为5%、10%、20%。利用高速剪切机进行乳化(20 000 r/min、2 min),制备得到不同含油量的蛋白乳液。
通过模拟体外小肠环境进行乳液脂肪消化研究。乳液用pH 7.0的Tris-HCl缓冲液稀释,使油相终质量分数为0.5%,微调pH值至7.0。取31 mL,37 ℃水浴振荡30 min,添加1.5 mL NaCl(5.625 mg/mL)、1 mL CaCl2(83.25 mg/mL)、2 mL胆盐(93.75 mg/mL)、2 mL胰脂肪酶(30 mg/mL)(以上溶液均用Tris-HCl缓冲液配制)。最终模拟肠液体积为37.5 mL,调节pH值至7.0。利用pH-stat全自动电位滴定仪进行体外消化实验,按式(2)计算游离脂肪酸的释放率,从而评价小肠中脂肪消化的程度[25]。
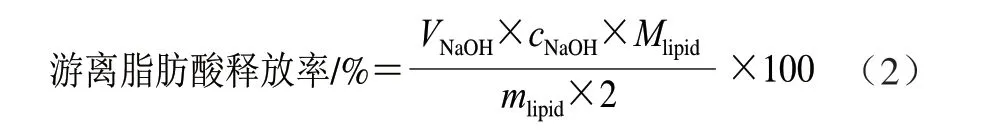
式中:VNaOH为中和游离脂肪酸所消耗的NaOH体积/L;cNaOH为所用的NaOH的浓度/(mol/L);Mlipid为中链甘油三酯摩尔质量(500 g/mol);mlipid为消化液中脂肪的总质量(150 mg)。
1.4 数据统计与分析
本实验的测试结果采用3 次测量取平均值的方法,以平均值±标准偏差表示。采用Origin 8.0、MATLAB 2016、Excel 2019软件进行数据统计和图表绘制。
2 结果与分析
2.1 蛋白聚集体形貌
蛋白质溶液在加热过程中蛋白质结构会发生变化,这一过程导致蛋白质分子间可发生相互作用,形成低聚物。当低聚体浓度超过一定浓度时,形成相对分散的初级聚集体。蛋白浓度越小、pH值越高,形成初级聚集体的粒径越小。如在pH 6时,可形成水力学半径约为150 nm近似球体的β-lg初级聚集物[3]。通过前期实验对不同pH值下的热处理蛋白质进行观察,发现pH 5.8条件下得到的纳米颗粒大小最为均一,且粒度较小,因此后期实验均使用在pH 5.8下制得纳米颗粒聚集体。图1A1、B1分别为β-lg及β-lg NP冻干后样品复溶的TEM结果。结合水合粒径分布图(图1A2、B2),可知β-lg为球状,粒径为(4.3±1.0)nm;而β-lg NP粒径在(160±10)nm左右,且单分散性良好,与文献[26]报道一致。
在低pH值下,对蛋白分子进行热处理,一般认为蛋白分子先水解为多肽,多肽再形成不同的次级结构(β-折叠),这些次级结构通过分子间相互作用组装形成纤维状聚集体[20]。所以可以通过控制pH值和温度来制备纤维状聚集体。图1C1、D1为β-lg F冻干前后TEM图,利用Fiber App软件对β-lg F冻干前后的纤维长度统计结果如图1C2、D2所示,冻干前,纤维长度分布为1 400~2 500 nm;而冻干后长度分布在100~700 nm之间,与冻干前相比,纤维长度明显减小。产生此现象的原因可能是冻干及其过程中的物理处理使蛋白质变得脆而易断导致[27]。通过前期实验对表面疏水性和表面电性测定[28],发现冻干前后表面性质无明显差异,因此为精确控制蛋白溶液浓度,本实验中所用聚集体均为冻干后样品。
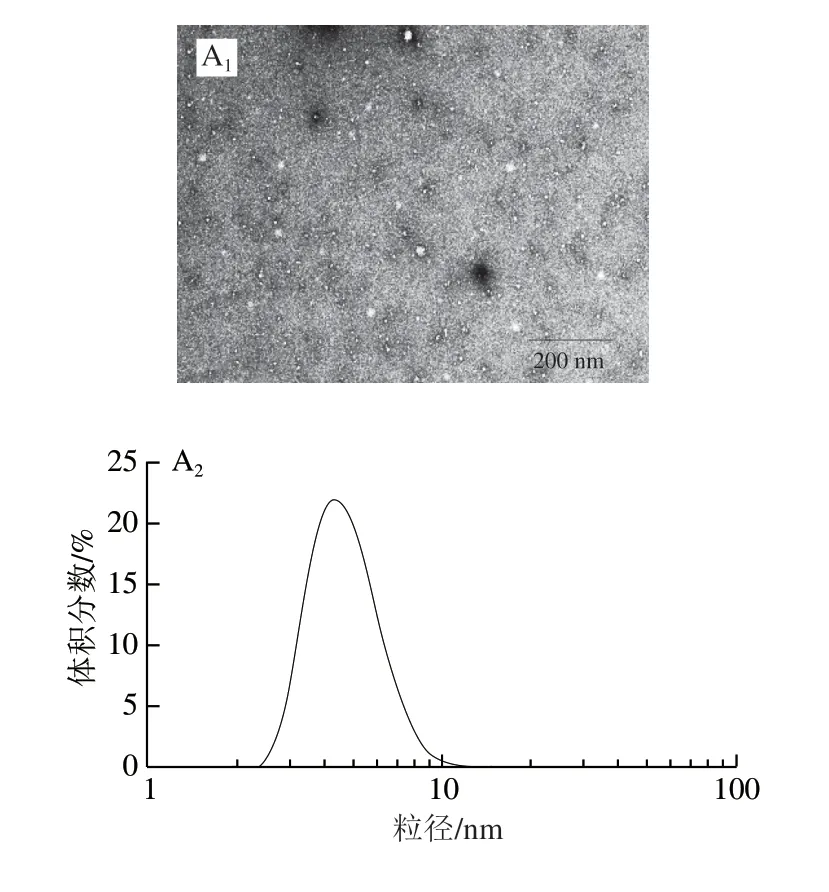
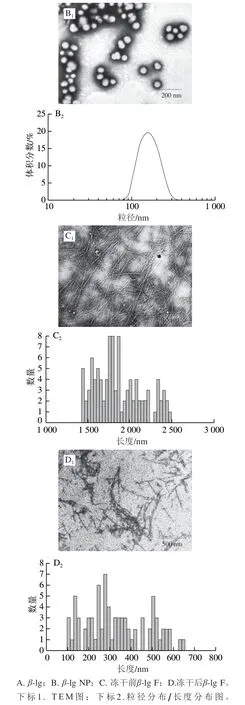
图 1 TEM图及粒径分布Fig. 1 Transmission electron microscope images and particle size distribution
2.2 蛋白聚集体的界面吸附
2.2.1 示踪粒子微流变学分析结果
在界面流变学研究中,在无蛋白的液-液界面上,示踪粒子在界面上的扩散系数D可以由改进的Stokes-Einstein方程(式(3))来描述[23]。

式中:kB为玻尔兹曼常数/(J/K);T为绝对温度/℃;η1和η1分别为上相和下相的黏度/(mPa·s);R为粒子半径/m;θ为粒子在油-水界面相对下相的接触角/(°);f(θ表示扩散系数取决于上下两相的黏度比和示踪粒子在界面上的接触角θ。实验中所用癸烷的黏度约为0.92 mPa·s,而所用蛋白缓冲液的黏度约为0.95 mPa·s,因此上相和下相对于粒子在界面上的黏度贡献可以认为相互抵消。同时根据前人测量的结果,聚苯乙烯荧光微球在癸烷-水界面上的接触角为90°[22],因此式中θ的影响被消除,公式(3)化简为标准的Stokes-Einstein方程(式(4))。

式中:η为蛋白溶液的黏度/(mPa·s)。此时界面示踪粒子的运动为布朗扩散运动,MSD可以从式(5)获得。

式中:τ表示扩散间隔时间/s。因此可以根据示踪粒子的运动计算得到无蛋白界面的扩散系数,从而得到下相黏度。采用1.3.3节的装置和加样方式测量了癸烷-水界面上示踪粒子的运动,得到扩散系数D0为0.566 μm2/s,相应的水相的黏度为0.771 mPa·s。测得的黏度略高于25 ℃纯水黏度,可能是由于粒子示踪实验中激光使得样品略加热。测量值在误差范围内,证明了方法的可行性。
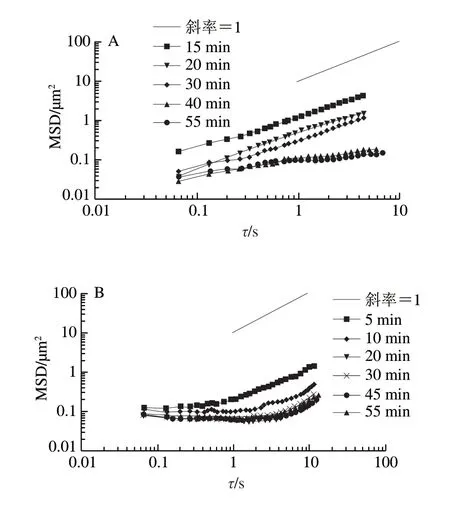
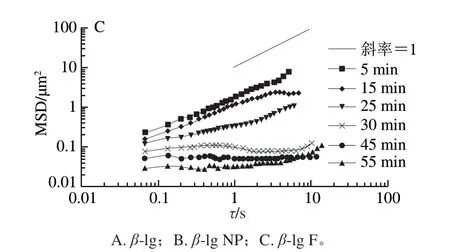
图 2 质量浓度均为110 μg/mL的不同形态蛋白聚集体在油-水界面吸附过程中示踪粒子的MSD曲线Fig. 2 MSD of tracer particles at the decane-water interface during the adsorption of proteins or protein aggregates at a concentration of 110 μg/mL
图2为110 μg/mL的β-lg、β-lg NP、β-lg F在油-水界面吸附过程中示踪粒子的MSD曲线。可以看出,随着吸附时间的延长,各样品MSD减小。对于不同流动行为,MSD与时间可以表示为幂指数关系,即<Δr2(t)>=4Dτa,a为幂指数。在双对数坐标中,MSD与时间关系曲线的斜率即为a。如果a等于1,即MSD随时间线性变化,说明粒子在牛顿流体中运动,处于自由扩散;如果a<1,表明粒子在具有黏弹性的物质中运动,处于亚扩散状态。当a趋近于0时,粒子被束缚在黏弹性较强的物质中,此时粒子的运动被严重限制,随时间的延长,MSD几乎是一个常数。本实验计算了不同质量浓度的β-lg、β-lg NP、β-lg F在不同吸附时间时油-水界面上示踪粒子MSD对时间的幂指数a(图3)。
由图3可以看出,随着时间的延长,对不同蛋白,a均减小,但不同形态蛋白降低快慢不同。如图3A所示,25 μg/mL下,对于β-lg界面,10 min时,a约为0.95左右,到50 min时,a约为0.75左右,示踪粒子运动被一定程度的限制;但对β-lg F界面,10 min时,a约为0.85,与β-lg接近,但50 min时,a约为0,示踪粒子运动被强烈限制;而对于β-lg NP界面,5 min时,a已经下降为0.56左右,30 min时a约为0。说明β-lg NP界面上示踪粒子的运动在蛋白吸附的初期就被限制,界面很快形成。这些结果说明不同形态的蛋白聚集体向界面扩散的速度不同,聚集体吸附速度快于β-lg,且β-lg NP最快。110 μg/mL和220 μg/mL蛋白聚集体同样显示出类似规律(图3B、C)。对于同一种蛋白,蛋白质量浓度越高,界面上示踪粒子的a下降越快,运动被限制得越明显,如β-lg在220 μg/mL下,40 min时,a为0,说明粒子运动被完全限制。通过公式可以计算出当蛋白质在油-水界面吸附成膜后蛋白膜的弹性模量(Gf)[17]。根据总MSD最后出现的平台值,计算得到220 μg/mLβ-lg的Gf为6.4×10-5mN/m(25、110 μg/mLβ-lg均未出现MSD平台值),以及25、110、220 μg/mL的β-lgNP的Gf分别为7.7×10-5、8.2×10-5、8.5×10-5mN/m,β-lg F的Gf分别为1.3×10-4、1.6×10-4、2.0×10-4mN/m。
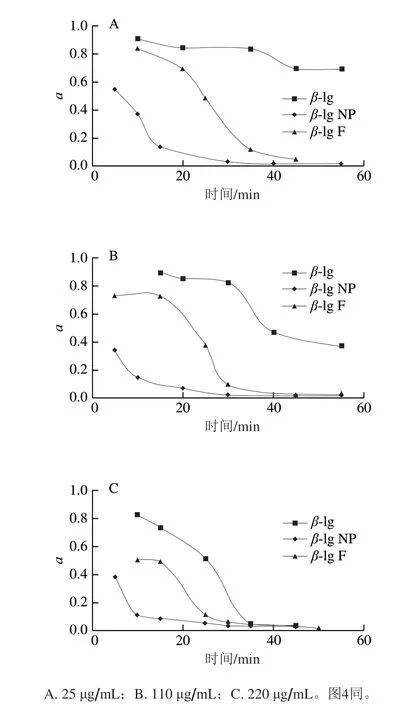
图 3 蛋白聚集体在油-水界面吸附过程中对数坐标中MSD斜率a随时间的变化Fig. 3 Change in the MSD’s slope in logarithmic coordinates during the adsorption process
2.2.2 界面膨胀流变学分析结果
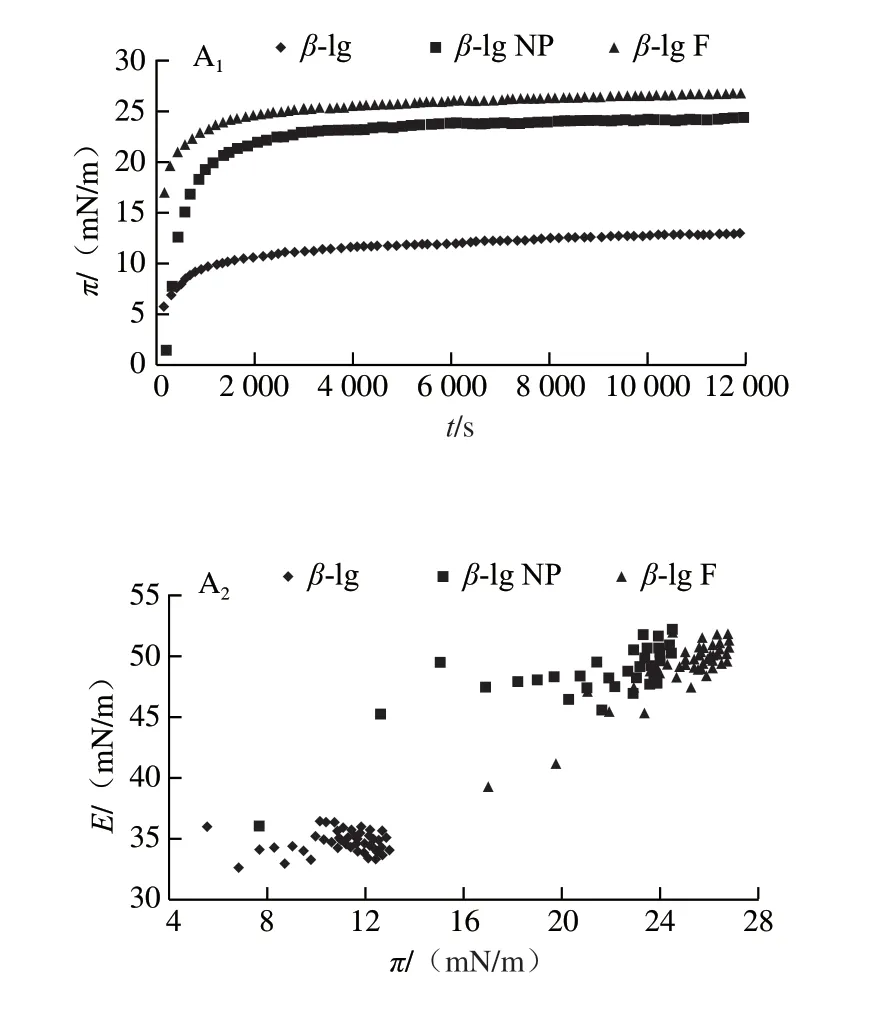
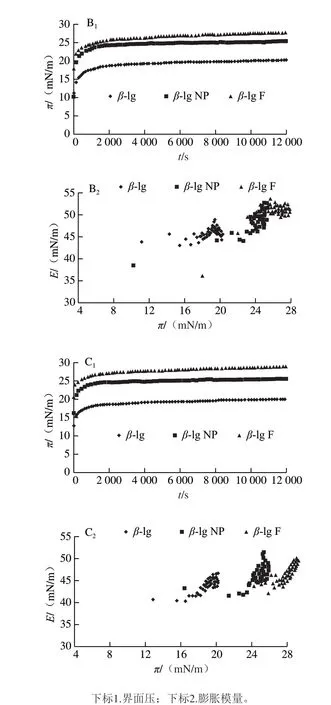
图 4 不同质量浓度不同形态蛋白向界面吸附的界面压及膨胀模量Fig. 4 Change in surface pressure with time and change in dilation module with surface pressure during protein adsorption
图4为不同质量浓度β-lg及聚集体β-lg NP和β-lg F向界面扩散过程中,界面压(π)及界面膨胀模量(E)的变化。可以看出在所考察的3 个质量浓度下,界面压均随时间延长快速增加,说明蛋白均向界面扩散。但不同质量浓度不同形态蛋白界面压强和膨胀模量变化不同。图5A、B分别为不同质量浓度的3 种形态蛋白的平衡后界面压及膨胀模量数值。从图5A可以看出,对于同种蛋白,随着质量浓度的增加,平衡后的界面压均增大;对于在相同质量浓度下的不同蛋白,β-lg F的界面压最大,其次是β-lg NP。从图5B可以看出,随着蛋白质量浓度的增加,界面蛋白膜的膨胀模量即黏弹性也随之增加;在相同质量浓度下,β-lg F的界面膜黏弹性最大,其次是β-lg NP,而β-lg的模量最小。
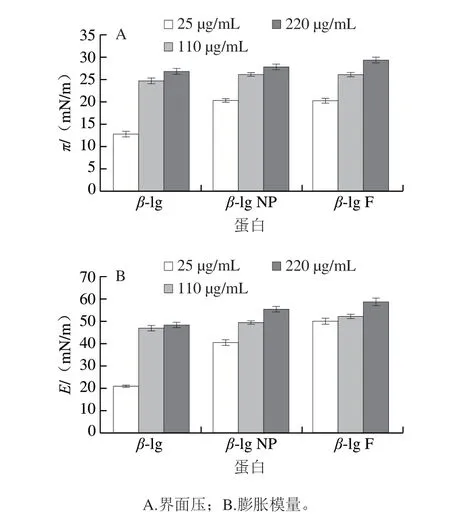
图 5 不同质量浓度不同形态蛋白平衡后界面压值及平衡后膨胀模量Fig. 5 Surface pressure values and dilation modulus after adsorption equilibrium for different β-lg protein aggregates at different concentrations
2.3 胆盐对界面蛋白的取代
2.3.1 示踪粒子微流变学结果
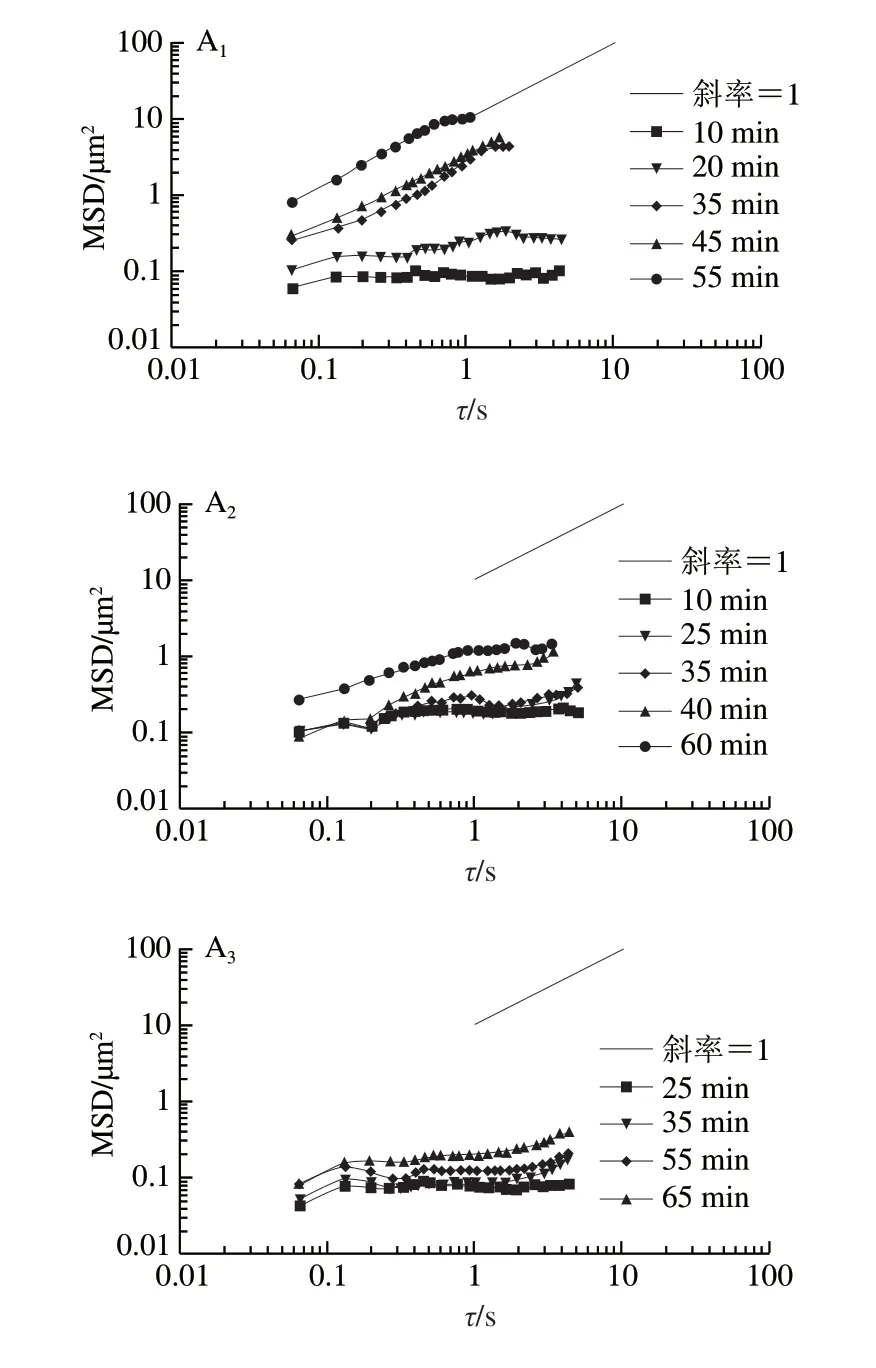
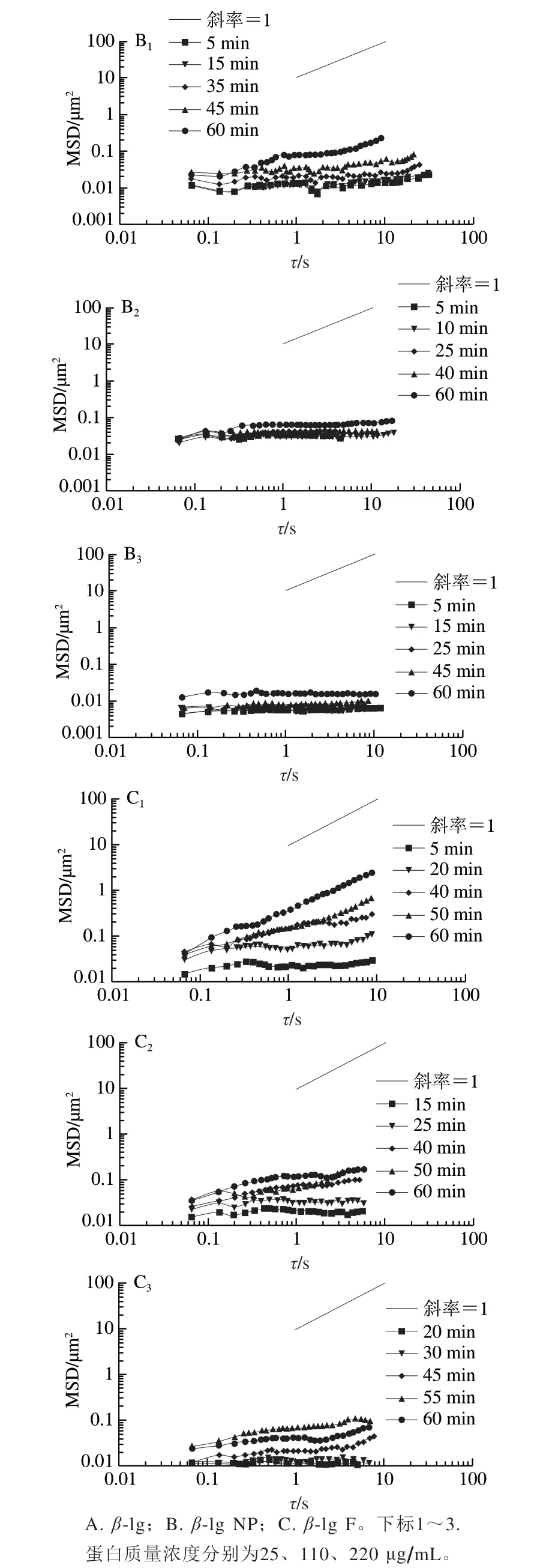
图 6 不同质量浓度蛋白及聚集体被胆盐取代过程中界面上示踪粒子整体平均MSD随时间的变化Fig. 6 MSDs of tracer particles at the decane-water interface during displacement of proteins by bile salts
图6分别为β-lg、β-lg NP、β-lg F在25、110、220 μg/mL下被胆盐取代过程中界面上示踪粒子整体平均MSD。在每个质量浓度下,随着胆盐取代时间的延长,MSD及其斜率a均逐渐增加,说明示踪粒子的运动限制逐渐缓解,原因可能是胆盐逐渐取代界面处蛋白,界面蛋白膜的网络结构逐渐打开。对于同一种蛋白,随着蛋白质量浓度的增加,MSD的斜率a变化减慢:如胆盐取代25 μg/mL的β-lg达到55 min时,a接近1;而胆盐取代110 μg/mLβ-lg达到60 min时,a约等于0.87;胆盐取代220 μg/mLβ-lg达到65 min时,a仅约等于0.56。这说明随着蛋白质量浓度的增加,胆盐取代界面蛋白的程度降低,蛋白界面膜仍保持一定的结构,示踪粒子的运动仍受到限制。
图6B1~B3为β-lg NP被胆盐取代的过程。可以看出,虽然胆盐对25 μg/mLβ-lg NP进行取代后,MSD的斜率a随着胆盐取代时间延长而逐渐变大,但相比于β-lg的a小;而110 μg/mL及220 μg/mL下,a变化不明显,在整个实验观察过程中均接近0,说明胆盐取代过程慢,甚至被抑制。
图6 C1~C3是β-l g F 被胆盐取代的过程。在25 μg/mL下,MSD的a随取代时间延长而增大,最终斜率也为1,但是MSD较β-lg小一个量级。随着蛋白质量浓度的增加,MSD的斜率a变化减慢,但最终斜率比β-lg的更小。
由上述结果可以看出,蛋白聚集体形成的界面比β-lg更难被胆盐取代,其中β-lg NP抗胆盐取代能力最强。原因可能是胆盐与蛋白及其聚集体在pH 7条件下都带负电荷,其中β-lg NP电荷量最大。当胆盐吸附至界面时,β-lg与β-lg NP蛋白分子之间存在较强的静电相互斥力,这也有可能减缓胆盐向界面吸附的速度。另外,球形颗粒状物质在界面有较高的能垒[29],因此从界面脱附很困难,这也抑制了胆盐的取代。
2.3.2 界面膨胀流变学分析结果
图7为不同质量浓度的β-lg、β-lg NP、β-lg F被胆盐取代后油-水界面压(π)的增加量(Δπ)及界面膨胀模量的减少量(ΔE)。图7A、B中,随着蛋白质量浓度的增加,对不同蛋白界面均有Δπ降低,ΔE减小;对比β-lg、β-lg NP、β-lg F,发现相同质量浓度下β-lg NP的Δπ最大,ΔE最小,而β-lg的ΔE最大的是β-lg。说明在相同的胆盐质量浓度下,随着蛋白质量浓度的增加,胆盐取代能力降低。综上,β-lg NP抵抗胆盐取代能力最强,其次是β-lg F,β-lg最弱。产生此结果的原因可能是因为聚集体疏水性较强形成的界面膜强度较大,抵抗胆盐取代能力较β-lg强。β-lg NP抵抗胆盐取代能力最强是因为β-lg NP被取代需要更大的自由能(ΔG)。经计算,得到β-lg NP的ΔG大约为830 kJ,β-lg F的ΔG大约为658 kJ[30]。
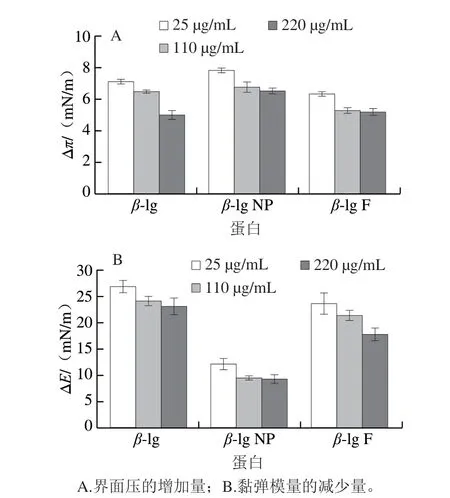
图 7 不同浓度蛋白被胆盐取代过程中界面压的增加量和膨胀模量的减少量Fig. 7 Increase in interfacial pressure and decrease in dilation modulus for different β-lg protein aggregates during bile salt displacement process
2.4 不同聚集形态蛋白质乳液的消化动力学分析结果
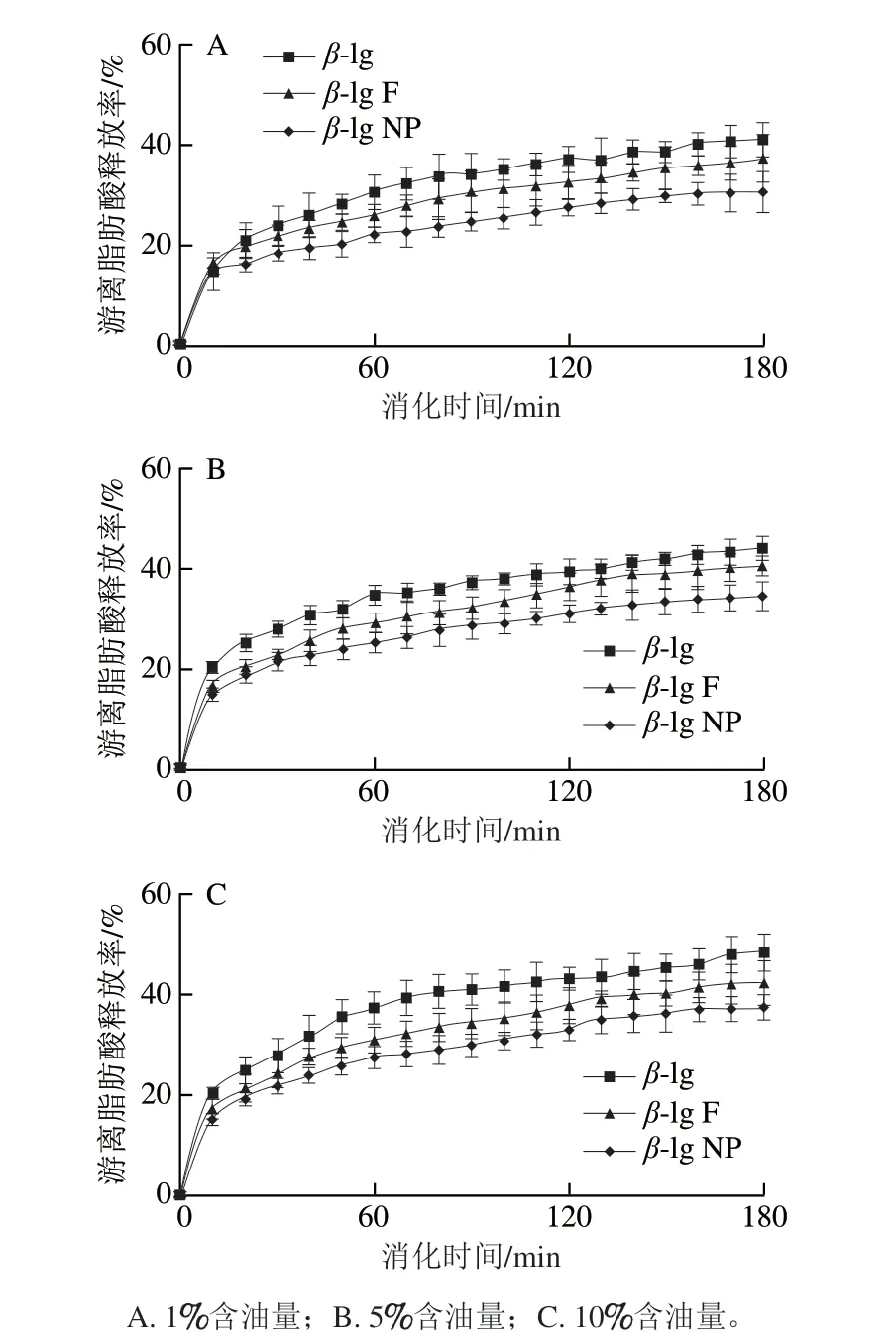
图 8 不同聚集形态蛋白质乳液脂肪消化过程中脂肪酸释放率变化Fig. 8 Free fatty acid release rate from emulsions stabilized by different β-lg protein aggregates during in vitro digestion
图8A、B、C分别为1%、5%、10%含油量条件下,不同形态蛋白质稳定的乳液在模拟肠道消化过程中脂肪酸的释放率。从图8中均可以看出,在模拟脂肪消化中,随着消化时间的延长,3 种蛋白质乳液的脂肪酸释放率逐渐增大。在相同含油量条件下,β-lg聚集体乳液体系脂肪消化速率低于天然蛋白乳液,其中β-lg NP乳液的脂肪消化速率最低。在脂肪消化过程中,胆盐与界面处蛋白质相结合,降低胆盐活动能力,进而影响脂肪消化速率。所以可以看出,在相同含油量的条件下,抗胆盐取代能力依次为β-lg NP>β-lg F>β-lg,与微流变实验结果一致。同时通过对不同形态蛋白质稳定的乳液的稳定性分析(结果此处未展示),发现蛋白聚集体乳液的稳定性较天然蛋白更强,这与微流变测得的模量和界面流变学测得的模量大小趋势也一致。
3 结 论
蛋白质动态吸附过程的瞬时信息捕捉存在一定困难,本实验利用示踪粒子微流变学观察到3 种形态蛋白质在油-水界面的实时微观流变信息。结果发现:1)3 种形态的蛋白质均可以向界面吸附,形成具有一定黏弹性的界面膜,且吸附成膜是一个动态过程;2)蛋白聚集体呈现出较强的界面活性,其在油-水界面吸附过程比β-lg快,β-lg NP吸附速度最快;3)蛋白聚集体形成的界面蛋白膜具有更强的黏弹性;4)β-lg NP抵抗胆盐取代能力最强,其次是β-lg F,β-lg最弱。微观流变学观察到的结果与膨胀流变学以及体外消化结果一致,说明了蛋白质聚集体在界面活性以及控制油脂消化方面的优势。
猜你喜欢
食品工业科技(2022年23期)2022-12-06
器官移植(2022年4期)2022-07-11
河南工业大学学报(自然科学版)(2021年6期)2022-01-26
化工科技(2021年5期)2021-11-24
中国饲料(2021年17期)2021-11-02
河南畜牧兽医(2021年10期)2021-01-05
天津中医药(2020年4期)2020-12-20
中国典型病例大全(2020年6期)2020-10-21
胃肠病学和肝病学杂志(2020年3期)2020-05-11
科技资讯(2019年23期)2019-11-11
