
COX-2/PGE2在肿瘤发生发展和重塑肿瘤微环境中的研究进展*
2020-09-19 01:56:08黎贵芸冯强胡雄边莉
中国肿瘤临床 2020年16期
黎贵芸 冯强 胡雄③ 边莉③
癌症相关炎症(cancer-related inflammation,CRI)在癌症的发生发展中发挥重要作用。甲状腺、肝脏、前列腺、胃肠道和胰腺等脏器的慢性炎症能增加患癌风险[1]。因此,CRI被认为是癌症的“推进器”,也是癌症复发的“导火线”[2]。生理情况下,急性炎症是机体的防御反应,旨在修复组织损伤和消除致病因子,多数情况下对机体是有利的。但是,当炎症调节失控时,有害因子长期存在,急性炎症转变为慢性炎症,可能会增加基因突变的几率、破坏适应性免疫,同时各种炎症介质过度表达,使炎症微环境向肿瘤微环境发展(tumor microenviroment,TME)。TME是由肿瘤细胞及周围基质细胞、免疫细胞、脉管系统、细胞外基质和细胞因子、生长因子、代谢物、微生物菌群等所形成的复杂网络[3]。无论在炎症微环境还是TME中,前列腺素E2(prostaglandin E2,PGE2)均是含量最丰富的类花生酸脂质,也是具有免疫调节功能的脂类代谢产物。研究表明,在结直肠癌、胃癌、胰腺癌、乳腺癌、肺癌等恶性肿瘤中环氧合酶-2(cyclo⁃oxygenase-2,COX-2)和微粒体前列腺素E 合酶-1(microsomal prostaglandin E synthase-1,mPGES1)及其代谢产物PGE2表达升高[4],提示PGE2与这些肿瘤的发生发展密切相关。流行病学研究还发现,抑制PGE2合成的非甾体抗炎药在肿瘤治疗中也取得一定成效[4-6]。
1 前列腺素E2的正常合成与代谢
当组织细胞受到各种非特异性刺激时,细胞膜上的磷脂在磷脂酶A2(phospholipase A2,PLA2)、COX-2、mPGES1等酶的作用下生成PGE2,PGE2也可以通过非酶促方式由PGH2代谢,但速度较慢[7]。细胞内合成的PGE2 被细胞膜上的多药耐药蛋白-4(multiple drug resistance-associated protein 4,MRP4)主动运输到细胞外,在局部环境通过自分泌或旁分泌与细胞表面的PGE2受体(E-prostanoid,EP)结合发挥功能[8]。细胞外的PGE2又可被前列腺素E2转运蛋白(prostaglandin transporter,PGT)重新转运回细胞内,15-羟基前列腺素脱氢酶(15-hydroxyprostaglandin dehydrogenase,15-PGDH)将其氧化为无活性的15-酮式前列腺素E2[9]。因此,15-PGDH、MRP4和PGT在调节细胞内外PGE2的动态平衡中也发挥重要作用。
2 COX-2/PGE2途径相关蛋白表达的临床意义
COX-2/PGE2 及合成途径中的调节蛋白在结直肠癌、肺癌、乳腺癌、头颈癌等恶性肿瘤中的表达量与肿瘤分级、转移、预后不良及降低肿瘤细胞对化疗药物敏感度相关[4,10]。Zhang等[11]在89例卵巢癌石蜡组织样本中发现COX-2的高表达与卵巢肿瘤的分级相关,在肺腺癌中COX-2 表达水平与淋巴结转移密切相关[12]。PGE2 代谢的关键酶15-PGDH 高表达与总生存期(overall survival,OS)和无复发生存期(re⁃currence-free survival,RFS)的改善相关[13]。研究还发现,在神经母细胞瘤、前列腺癌、胰腺癌、上皮性卵巢癌和急性髓性白血病等肿瘤中,PGE2 胞外转运蛋白MRP4高表达与预后不良相关[14-19],基底样和HER2过表达型乳腺癌中MRP4 的表达水平与乳腺癌的侵袭性相关[13]。在三阴性乳腺癌中COX-2High、MRP4High、PGTLow、15-PGDHLow的表达模式利于TME中PGE2 的积累,这可能是导致三阴性乳腺癌预后不良的重要原因之一[13]。
3 COX-2/PGE2途径在TME中的作用及机制
3.1 微环境中COX-2/PGE2合成的调控机制
大多数肿瘤中的COX-2/PGE2处于高水平状态,受细胞内外多种信号转导途径及因子的调节。据文献报道,激活癌细胞的Braf/MEK[20]、磷脂酰肌醇-3 激酶(phosphatidylinositol 3-kinase,PI3K)信号途径可诱导COX-2 表达上调[6],EPHA2/TGF-β/SMAD/PTGS2[21]、PGE2/RhoC/PTGS2的正向调节环增加癌细胞COX-2/PGE2的表达量[22]。肺癌细胞中N-myc与STAT互作因子(N-myc and STAT interactor,NMI)通过抑制转录共激活因子P300介导的核因子-κB(nuclear factor-kappa B,NF-κB)P50/P65乙酰化来抑制COX-2/PGE2表达[23]。COX-2/PGE2途径除了受癌细胞内部信号通路活化的影响,转录后调控因子微小RNA(microRNA,miR)在PGE2的表达分泌中也十分重要。研究表明,非小细胞肺癌表达miR-574-5p,其GU富集元件(GU-rich elements,GRE)可作为RNA结合蛋白1(RNA-binding protein CUG RNA-binding protein 1,CUGBP1)的诱饵,阻止其与靶基因mPGES-1的3'端非翻译区(3'untranslated regions,3'UTR)结合,导致mPGES-1的3'UTR发生可变剪接生成新的同工型mPGES-1,增加mPGES-1蛋白表达,促进PGE2的表达[24]。此外,COX-2蛋白去糖基化修饰或细胞处于缺氧、垂死状态下也可促进PGE2释放[25-26]。研究还发现,药物也能影响肿瘤细胞COX-2的表达,COX-2抑制剂塞来昔布可反常增加癌细胞COX-2的表达,并通过外泌体将COX-2递送到肿瘤周围的单核细胞中,单核细胞吸收外泌体中COX-2后,增加PGE2的分泌[27]。阿片类药物结合乳腺癌细胞δ阿片受体可激活PI3K/AKT信号通路,上调COX-2的表达[28]。
在TME中,间质细胞、免疫细胞等细胞也存在多种COX-2/PGE2 过表达的调控机制。髓源抑制细胞(myeloid-derived suppressor cells,MDSC)中受体相互作用蛋白激酶3(receptor-interacting protein kinase 3,RIPK3)的下调,增强了NF-κB的转录、上调COX-2的表达和PGE2分泌[29]。在巨噬细胞中,转录因子YY1(yin yang1)与COX-2的启动子区结合可增加COX-2的转录活性[30]。在肿瘤相关成纤维细胞(cancer-associated fibroblasts,CAFs)中组蛋白脱乙酰基酶6(histone deacetylases 6,HDAC6)通过信号转导和转录激活子3(signal transducer and activator of transcription 3,STAT3)途径诱导COX-2/PGE2过表达[31],见图1。
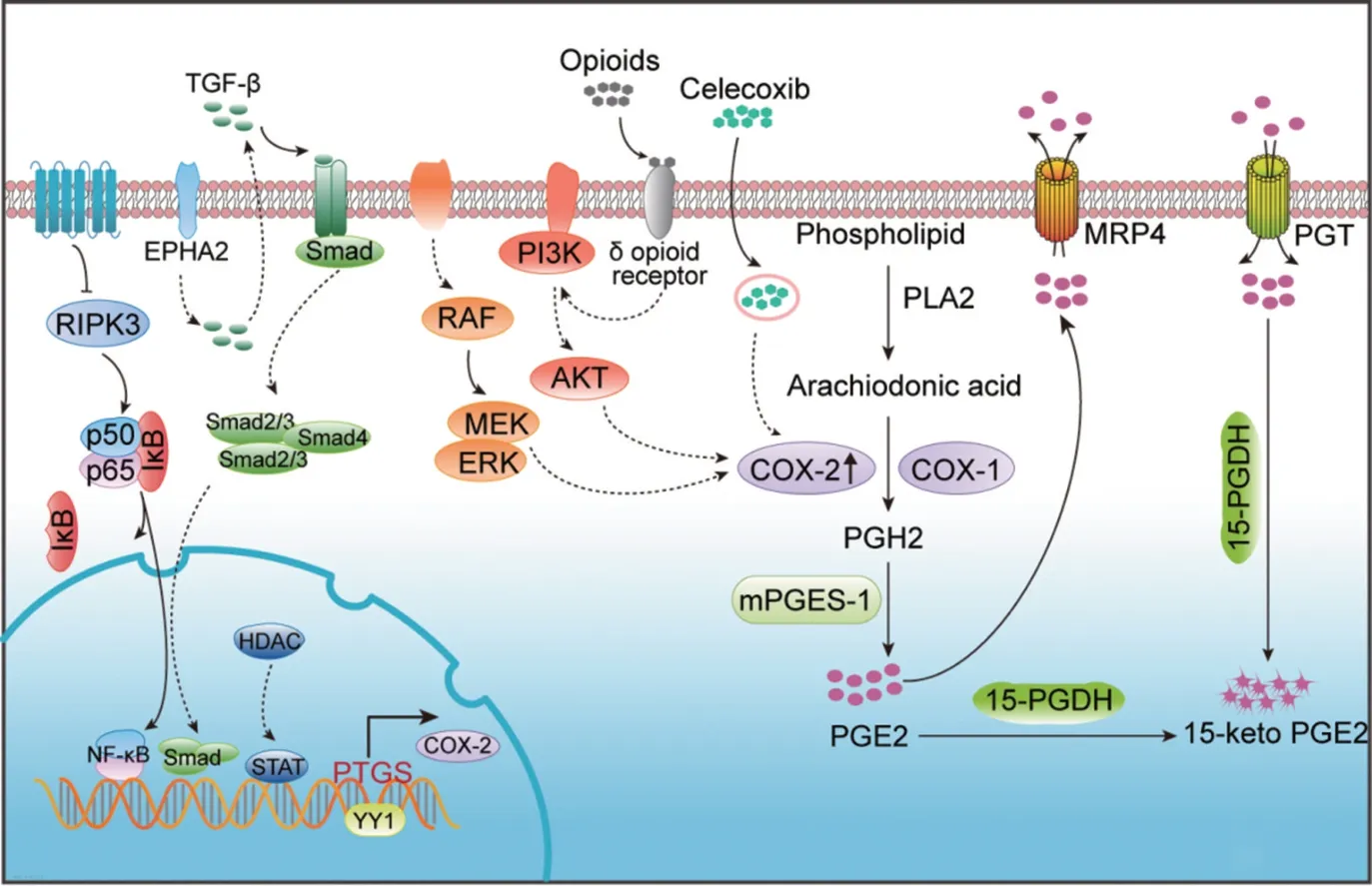
▶图1 细胞中COX-2/PGE2 合成的调控机制[31]
3.2 COX-2/PGE2直接调控肿瘤发生发展的机制
在TME中,肿瘤细胞及周围免疫细胞均可过表达COX-2,通过自分泌或旁分泌PGE2作用于肿瘤细胞和周围免疫细胞,直接或间接调控肿瘤的恶性生物学行为。在结直肠癌前病变APCmin/+小鼠模型中,PGE2通过结合受体EP1 和EP2 促进cAMP 反应元件结合蛋白(cAMP-response element binding protein,CREB)调控的转录共激活因子1(CREB regulated transcription coact⁃ivator 1,CRTC1)去磷酸化和核转移,增强CRTC1的转录活性,导致散发性或结肠炎相关的结肠癌发生[32]。外源性诱导人口腔上皮角质细胞(human oral kerati⁃nocyte,HOK)过表达COX-2/PGE2,可促进HOK增殖,同时上调口腔癌相关基因cyclin D1,活化NF-κB和抑制肿瘤坏死因子(tumor necrosis factor,TNF)信号传导途径中的相关基因JUN、Toll样受体4(Toll like receptor 4,TLR4)、CXC趋化因子1(C-X-C motif chemokine 1,CXCL1)、白血病抑制因子(leukemia inhibitory factor,LIF)、CXCL3、肿瘤坏死因子受体超家族1β(tumor ne⁃crosis factor receptor superfamily 1β,TNFRSF1β)和白细胞介素1β(Interleukin-1β,IL-1β),下调多种肿瘤抑制因子1(multiple tumor suppressor 1,MTS1)的表达,最终促使HOK细胞的恶性转化[33]。COX-2过表达还可驱动癌基因KRAS,从而激活丝裂原活化蛋白激酶(mitogenactivated protein kinase,MAPK)途径促进肺癌的发生[34]。上述研究表明,TME中的非转化细胞或癌前病变细胞均高表达COX-2/PGE2,以促进慢性炎症疾病向肿瘤发展,提示此类细胞过表达COX-2/PGE2可能是肿瘤发生的早期分子事件。
COX-2/PGE2 途径还能通过重编程肿瘤细胞生长参数、影响肿瘤细胞表型,为肿瘤的增殖、侵袭、转移提供条件。新近研究报道,卵巢癌细胞高表达COX-2、分泌PGE2,与其受体结合后上调NF-κB P65的磷酸化水平,增强卵巢癌细胞的增殖能力[11]。PGE2 与受体EP3 结合后激活SRC 家族激酶(SRC family kinases,SFK),诱导去整合素和金属蛋白酶(a disintegrin and metalloprotease,ADAMs)活化,ADAMs又反过来介导EGFR的配体两性调节蛋白(amphireg⁃ulin,AREG)和表皮调节素(epiregulin,EREG)的脱落,继而使EGFR 从细胞膜释放向核内转移,进入细胞核的EGFR 可延长诱导型一氧化氮合酶(inducible nitric oxide synthase,iNOS)、c-myc、cyclin D1、PTGS2等基因的表达,促进肿瘤细胞增殖[35-36]。垂死肿瘤细胞释放的PGE2作为抑制型损害相关分子模式(dam⁃age associated molecular patterns,DAMPs)抵消激活型DAMPs 的作用,抑制TNF-α 等关键性炎症细胞因子的表达,阻止炎症细胞因子继续对存活肿瘤细胞的攻击,表明PGE2可重塑TME来辅助存活肿瘤细胞增殖[26]。COX-2/PGE2 还可通过激活PI3K 和细胞外信号调节激酶(extracellular regulated protein kinases,ERK)途径诱导肿瘤细胞表达血管内皮生长因子受体(vascular endothelial growth factor receptor,VEG⁃FR)[37],增强癌细胞的迁移能力;通过激活STAT3、AKT/NOTCH/WNT 等途径或介导CXCR2 信号下调PI3K-P85α 促进癌细胞发生上皮间质转化(epitheli⁃al-mesenchymal transition,EMT)[38-39];还能激活PKC/MAPK/E2F-1/FoxC2信号通路上调癌细胞中β1-整合素的表达,促进癌细胞侵袭[12]。
3.3 COX-2/PGE2重塑TME间接调控肿瘤发生发展的机制
3.3.1 抑制淋巴细胞抗肿瘤能力 高水平的COX-2/PGE2 不仅能直接调控肿瘤细胞的恶性生物学行为,还可通过阻碍各种免疫细胞中的促炎反应并介导TME 的重编程,使TME 处于免疫抑制状态[40]。研究表明,PGE2诱导CD8+T细胞激活cAMP-PKA途径,使参与维持线粒体和细胞质之间氧化还原平衡的苹果酸-天冬氨酸穿梭(malate-aspartate shuttle,MAS)系统严重受损,下调MAS 中的天冬氨酸及多种酶和中间代谢物含量,致使CD8+T 细胞生长停滞[41-42],这可能是PGE2 降低TME 中CD8+T 细胞浸润数量的关键机制之一。另外,COX-2 编码基因PTGS2 的mRNA水平与CD8A、CD8B 转录水平及CXCL10、CXCL9 的表达呈负相关[20],影响CD8+T 细胞的募集。PGE2 还能抑制NK 细胞产生趋化因子CCL5和XCL1,同时下调cDC1细胞中趋化因子受体CCR5和XCR1的表达,抑制细胞毒性T 淋巴、NK 细胞-cDC1 细胞的募集作用及抗肿瘤能力[41]。上述研究表明,肿瘤微环境中高浓度的PGE2可抑制CD8+T细胞、NK-cDC1细胞的募集、增殖,以降低肿瘤周围杀伤细胞的浸润丰度,阻断该途径可能重新增加TME 中细胞毒性T 淋巴细胞或其他杀伤细胞浸润数量,提高免疫系统的抗肿瘤能力。研究还发现,COX-2/PGE2途径可诱导肿瘤细胞过表达程序性死亡蛋白配体(programmed cell death protein ligand 1,PD-L1),过表达的PD-L1 与T细胞表面的程序性死亡蛋白受体(programmed cell death protein 1,PD-1)结合后抑制T 细胞的杀伤作用,介导肿瘤细胞发生免疫逃逸。转录因子YY1 和TGF-β 刺激肿瘤细胞表达COX-2/PGE2 通过诱导癌细胞AKT 的磷酸化来激活PI3K/AKT 途径,进而激活mTOR,促进肿瘤细胞中PD-L1的表达[43]。而抑制转录因子YY1、EGFR、TGF-β 的表达时,COX-2/PGE2的表达也被抑制,进而下调PD-L1的表达[30,44]。
3.3.2 影响巨噬细胞活化状态 TME 中,COX-2/PGE2 还能促进MDSC 和巨噬细胞的募集、迁移和增殖,但是对MDSC和巨噬细胞功能极化的影响尚存争议。经典活化型巨噬细胞M1 和替代活化型巨噬细胞M2分别具有促炎性和抗炎性,NF-κB介导巨噬细胞向M1 型极化[45],恢复巨噬细胞的促炎功能,而NF-κB 又是COX-2 的反式激活因子,有助于激活肿瘤细胞和巨噬细胞中COX-2 的表达[46],二者具有相同的信号分子,COX-2参与调节巨噬细胞向M1型极化。有研究认为,COX-2是促进巨噬细胞向M2型极化的调节剂,MDSC 中RIPK3 下调促进了NF-κB/COX-2/PGE2 轴的活化,产生大量的PGE2,促进MD⁃SC 向M2 型巨噬细胞发生极化,同时PGE2 还可进一步降低RIPK3的水平,形成信号正反馈环以进一步促进MDSC的免疫抑制活性和致癌作用[29]。PGE2转运蛋白表达异常可升高细胞外PGE2的水平,肿瘤细胞条件培养基中抑制髓源细胞PGT 和15-PDGH 的表达,增强了MRP4的表达,促进髓源细胞转化为M2型巨噬细胞[47]。在结直肠癌前病变APCmin/+小鼠模型中,COX-2+的肿瘤相关巨噬细胞高表达精氨酸酶(ar⁃ginase-1,Arg-1)、不表达iNOS[48]。也许COX-2 并非巨噬细胞极化的特异性决定因素,但是其可以充当巨噬细胞前期活化的促进剂。
3.3.3 促进血管、淋巴管生成 COX-2/PGE2途径不仅可以通过调控癌细胞自身VEGFR表达促进肿瘤进展,还刺激血管内皮生长因子(vascular endothelial growth factor,VEGF)合成,诱导内皮细胞、CAFs的迁移和分化作用来触发血管内皮细胞和淋巴管内皮细胞萌发,促进肿瘤细胞血管和淋巴管生成[49]。在胶质瘤细胞中阻断COX-2和mPGES-1的作用可抑制AKT磷酸化,通过miR-194-5p和长链非编码RNA(long non-coding RNA,lncRNA)NEAT1的内源性竞争作用降低VEGF、成纤维细胞生长因子-2(fibroblast growth factor-2,FGF-2)、转化生长因子-β(transforming growth factor-β,TGF-β)的产生,以阻碍肿瘤血管新生[50]。基质成纤维细胞和成骨细胞表达COX-2和mPGES-1上调PGE2,PGE2与EP4受体结合诱导VEGF、碱性成纤维生长因子(basic fi⁃broblast growth factor,bFGF)、NF-κB受体活化因子配体(receptor activator of nuclear factor-κB ligand,RANKL)表达,促进肿瘤血管生成,并诱导破骨细胞活化,产生溶骨作用,为癌细胞发生骨转移提供条件[51],见图2。
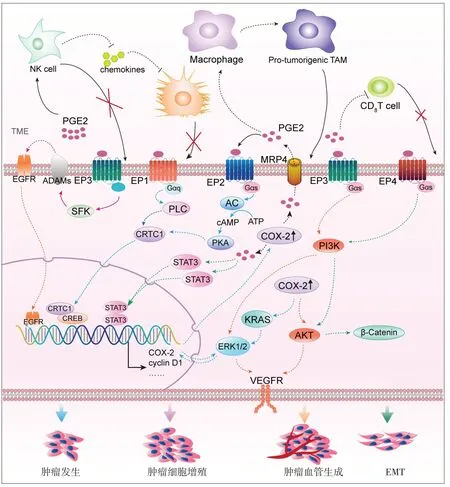
▶图2 COX-2/PGE2 途径在TME中的作用及调控机制[51]
3.3.4 对CAFs的影响COX-2/PGE2促进CAFs 表达miR-335介导PTEN蛋白下调,进而促进CAFs 向衰老相关分泌表型(senescence associated secretory phenotype,SASP)发展,该表型的CAFs细胞是癌细胞运动的诱导剂[52]。TGF-β 诱导锌指蛋白SNAIL1 表达触发CAFs产生PGE2,进而增加乳腺癌细胞的侵袭性[53]。小鼠前列腺癌细胞分泌的PGE2,通过激活与催乳素基因PRL 调控序列结合的孤儿核受体4A-类维生素X 受体(orphan nuclear receptor 4A-retinoid X receptor,RXR),诱导肿瘤邻近基质CAFs产生和分泌催乳素,催乳素反过来结合肿瘤细胞上的催乳素受体激活信号通路,从而促进肿瘤增殖[54]。
3.3.5 对干细胞的影响 此外,COX-2/PGE2还能激活肿瘤干细胞(cancer stem cells,CSCs)的干性,Kurto⁃va 等[55]在膀胱癌患者中发现,化疗诱导肿瘤细胞凋亡,随后释放PGE2,共定位技术显示PGE2 主要分布于CSCs周围,用外源性PGE2与经化疗处理的膀胱癌细胞上清分别刺激肿瘤干细胞,二者均能增强CSCs的成球能力。PGE2 与其受体EP4 结合激活PI3K 和MAPK途径导致转录因子NF-κB活化,诱导小鼠结直肠癌CSCs 的增殖并促进肿瘤细胞向肝脏转移[56]。PGE2 通过下调E-钙黏蛋白的表达,促进β-catenin的释放和核易位,核β-catenin 与TCF/LEF 协同激活干细胞多能性关键基因NANOG、Oct4 和Sox2 的表达,恢复CSCs 的干性[57]。另外,caspase 募集功能域的凋亡抑制因子(apoptosis repressor with caspase re⁃cruitment domain,ARC)调控癌细胞产生白细胞介素-1β(interleukin-1β,IL-1β),IL-1β诱导间充质干细胞(mesenchymal stromal cells,MSCs)表达COX-2、分泌PGE2,PGE2 又以旁分泌的方式作用肿瘤细胞激活β-catenin 信号,刺激癌细胞获得CSCs 表型;同时βcatenin又能上调ARC的表达,癌细胞则在MSCs依赖的ARC/IL-1β/Cox-2/PGE2/β-catenin 信号回路环的保护作用下抵抗化疗[58]。上述研究证实,COX-2/PGE2 途径可通过β-catenin 信号通路激活CSCs 表型、诱导CSCs增殖来保护肿瘤细胞抵抗化疗,促进肿瘤的发生、进展和转移。
4 结语与展望
免疫疗法单药治疗的超进展、肿瘤治疗耐药性是当前肿瘤治疗的难题之一,联合用药是缓解这一难题最快速有效的方法之一。有报道使用COX-2抑制剂依托度酸可抑制M2型巨噬细胞的特性,降低小鼠乳腺癌的肺转移率[59],COX-2 特异性抑制剂塞来昔布可增强胰腺癌对免疫检查点抑制剂和CD40 激动剂的敏感性[60]。COX-2 shRNA 抑制COX-2 的表达逆转了乳腺癌细胞对吉西他滨和紫杉醇化疗耐药性[61]。另外,肿瘤细胞及周围间质细胞接收自身PGE2 信号刺激的同时也接收周围细胞旁分泌的PGE2,促使TME长期处于免疫抑制状态,了解COX-2/PGE2 途径上下游的调控机制及分子事件,开发COX-2/PGE2途径精准抑制剂,打破该途径造成的免疫抑制状态,再联合使用免疫靶点抑制剂或其他化疗药物,可能会取得较好的抗肿瘤效果。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10 09:15:38
作文成功之路·小学版(2020年6期)2020-07-27 01:48:28
中学生数理化·七年级数学人教版(2019年10期)2019-11-25 07:33:58
奥秘(创新大赛)(2019年9期)2019-10-09 02:03:56
小哥白尼(趣味科学)(2019年1期)2019-04-12 00:23:56
中学生数理化·高一版(2018年9期)2018-10-09 06:46:50
湖南教育·C版(2018年3期)2018-06-05 16:54:36
奥秘(2017年5期)2017-07-05 11:09:30
环境与生活(2016年6期)2016-02-27 13:47:10
无机化学学报(2014年10期)2014-02-28 17:33:13
