
GC-MS法测定甲磺酸中3种甲磺酸烷基酯类遗传毒性杂质
2020-09-14 02:09陈忆铃冯江江杨海雪时雅萍李龙囡
中国药科大学学报 2020年4期
陈忆铃,冯江江,杨海雪,时雅萍,李龙囡,冯 芳*
(1中国药科大学药物分析系,南京211198;2无锡市药品检验所,无锡214028;3扬子江药业集团南京海陵药业有限公司,南京210049)
甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯是DNA 反应活性物质,在极低水平下仍存在致突变/致癌风险[1-4],为此,全球药品监督管理部门高度重视,纷纷要求相关企业在药物研发、生产过程中对其严格控制[5-7],国家药典委员会在即将执行的2020 版《中华人民共和国药典》中新增了“遗传毒性杂质控制指导原则”[8]。
甲磺酸盐类药物中甲磺酸烷基酯杂质可能来源于副反应和起始物料甲磺酸。Teasdale 等[9-10]通过实验研究,提出了在甲磺酸盐合成过程中最大限度的减少或消除甲磺酸烷基酯的策略。除酸碱化学计量比、酸度、温度、水分含量及反应时间等影响外,起始物料甲磺酸的质量十分关键。由于甲磺酸成盐反应主要发生在药物合成的最终阶段[11-12],成盐过程中甲磺酸含有的甲磺酸烷基酯杂质很可能被带入药物终产物中,带来安全隐患。因此,对甲磺酸中可能存在的甲磺酸烷基酯进行分析监测,对于甲磺酸盐类药物的安全和质量控制十分重要。
有关甲磺酸中甲磺酸烷基酯类杂质检测的报道不多。Ramjit等[13]以乙腈为溶剂,采用直接进样GC-MS 法测定了甲磺酸中甲磺酸甲酯、甲磺酸乙酯、甲磺酸正丙酯及甲磺酸正丁酯,虽然方法简单,便于操作,但样本基质容易在气相进样口造成污染。Zhou 等[14]尝试采用柱前衍生化-高效液相色谱法测定甲磺酸中甲磺酸烷基酯。由于所用衍生化试剂的亲核性较差,该法仅能测定供试品中的甲磺酸甲酯和甲磺酸乙酯,且每个样品分析需时30 min。欧洲药典采用二氯甲烷液液萃取后GC-MS 法分析测定甲磺酸中的甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯[15],方法表观简单,但实际应用时出现回收率偏低的问题。本研究在欧洲药典方法的基础上,通过改变对照品溶液的配制方法提高方法的定量准确度,并考察了质谱定量离子及检测方式、毛细管色谱柱及升温程序、进样方式及进样参数等对方法专属性和灵敏度的影响,进一步通过比较单点校正法与标准曲线法的准确度,确立了内标定量方法,并将该方法应用于不同来源的甲磺酸质量控制与监测。
1 材 料
1.1 仪 器
GC-MS-QP2020气相色谱-质谱联用色谱仪,配有AOC-20i 自动进样器,GC-MS solution 4.45 版工作站(日本Shimadzu 公司);BSA124S 型电子天平、BT25S型电子天平(德国Sartorius公司)。
1.2 试 剂
甲磺酸甲酯(99%,批号:A0356876)、甲磺酸乙酯(99%,批号:A0341901)、甲磺酸异丙酯(99%,批号:A0375206),均购自美国Acros Organics 公司;甲磺酸正丁酯(98%,批号:AK114028,美国Ark Pharm 公司);二氯甲烷(色谱/光谱级,批号:19045160,美国Tedia 公司);甲磺酸(≥98%,批号:10210923,美国Alfa Aesar 公司);甲磺酸(99.5%,批号:F1801073、D1919052,阿拉丁试剂有限公司);其余均为市售分析纯试剂。
2 方 法
2.1 色谱与质谱条件
2.1.1 色谱条件 Agilent HP-1MS 毛细管柱(固定液为100%二甲基聚硅氧烷,30 m × 0.32 mm,1 µm);进样口温度220 ℃;柱温:初始温度为55 ℃,维持1 min,10 ℃/min 升温至95 ℃,维持2 min;再以10 ℃/min 升温至135 ℃,维持2 min;载气:高纯氦气;流速为2 mL/min;不分流高压进样:压力250 kPa,时间0.5 min;进样量:2µL。
2.1.2 质谱条件 电子轰击源(EI),离子源温度:230 ℃;接口温度280 ℃;四极杆质量分析器,选择离子监测(SIM)模式。检测离子、检测时间窗信息见表1。
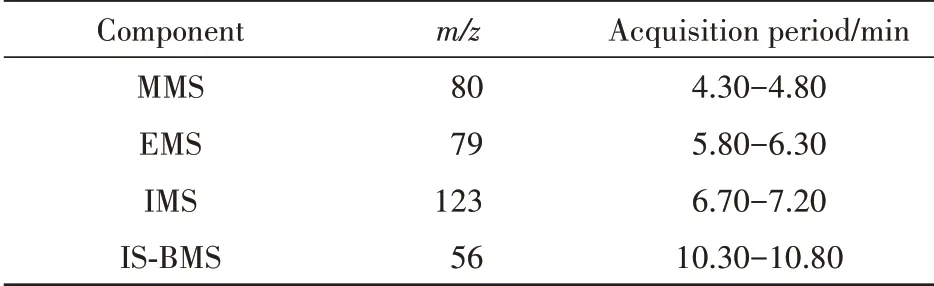
Table 1 SIM mode of components to be tested
2.2 溶液制备
2.2.1 空白溶液 于分液漏斗中精密加入水5 mL及二氯甲烷5 mL,振摇提取,静置使分层;取下层有机相加无水硫酸钠0.7 g,振摇,静置5 min,取上清液,即得。
2.2.2 内标溶液 量取甲磺酸正丁酯7 µL,置10 mL 量瓶中,加二氯甲烷稀释至刻度,摇匀,作为内标贮备液;精密量取内标贮备液5µL,置100 mL量瓶中,加二氯甲烷稀释至刻度,摇匀,即得。
2.2.3 混合对照品溶液 精密称取甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯各25 mg,置25 mL量瓶中,加内标溶液稀释至刻度,摇匀,作为混合对照品贮备液;精密量取混合对照品贮备溶液74 µL,置10 mL 量瓶中,加内标溶液稀释至刻度,摇匀,作为混合对照品中间贮备液;精密量取上述溶液100µL,置10 mL量瓶中,加内标溶液稀释至刻度,摇匀,备用;于分液漏斗中精密加入5 mL水及上述溶液5 mL,振摇提取,静置使分层;取下层有机相加无水硫酸钠0.7 g,振摇,静置5 min,取上清液,即得。
2.2.4 供试品溶液 取供试品0.37 g,精密称定,置分液漏斗中,精密加入水5 mL,摇匀,冷却至室温;精密加入内标溶液5 mL,振摇提取,静置使分层;取下层有机相加无水硫酸钠0.7 g,振摇,静置5 min,取上清液,即得。
3 结 果
3.1 专属性考察
取空白溶液、混合对照品溶液、供试品溶液(≥98%,批号:10210923,美国Alfa Aesar 公司),按“2.1”项下条件进样分析,记录色谱图(见图1)。结果表明,甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯和甲磺酸正丁酯的保留时间分别为4.63、5.99、6.89、10.46 min,空白溶液(图1-A)不干扰测定,各相邻峰之间分离度良好(图1-B、1-C),方法专属性良好。
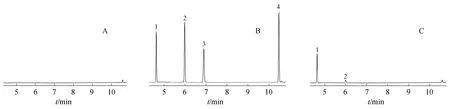
Figure 1 Characteristic GC-MS chromatograms of blank solution(A),standard solution(B)and test solution(C)
3.2 线性关系考察
取混合对照品中间贮备液适量加内标溶液稀释至刻度制得质量浓度约为37,74,370,740,1 480 ng/mL 对照品溶液,经萃取-干燥处理后制得系列标准线性溶液,按“2.1”项下条件进样分析,以各待测物与内标峰面积比为纵坐标,浓度为横坐标进行线性回归,各甲磺酸酯在37~1 480 ng/mL范围内线性关系良好,结果见表2。

Table 2 Result of linearity test of three alkyl methanesulfonates
3.3 定量限和检测限
MMS、EMS、IMS的定量限分别为36.75、35.49和36.14 ng/mL,定量限精密度分别为1.25%、0.68%和0.37%,定量限准确度分别为105.73%、105.32%、101.53%。以信噪比约为3∶1 的相应浓度作为检测限,MMS、EMS、IMS 的检测限分别为7.35、7.10和7.23 ng/mL。
3.4 进样精密度
取混合对照品溶液,连续进样5 次,MMS、EMS、IMS 与内标峰面积比的RSD 分别为0.51%、0.43%和0.27%,仪器进样精密度良好。
3.5 回收率
取甲磺酸样本(批号:10210923)共9 份,每份0.37 g,精密称定,加水5 mL 稀释,再分别加入质量浓度为37、370 和1 110 ng/mL 对照品溶液5 mL,经提取干燥制得低、中、高3 种质量浓度的加样回收率溶液,每个质量浓度水平各3 份,分别进样分析。另取混合对照品溶液,同法测定。回收率试验结果见表3,该方法准确度良好。

Table 3 Recovery of three alkyl methanesulfonates
3.6 重复性
按“3.5”项下方法制备加标质量浓度分别为37、370 和1 110 ng/mL 的供试品溶液,分别进样分析,记录峰面积,计算得各浓度水平下各甲磺酸酯峰面积的RSD 均不超过2.78%,该方法重复性良好。
3.7 稳定性考察
取混合对照品中间贮备液适量加内标溶液稀释至刻度制得质量浓度约为74、370、740 ng/mL 对照品溶液,经萃取-干燥处理后,于室温(25 ℃)下放置0 h 和8 h 后连续进样分析(n=3),结果表明,各浓度水平下各甲磺酸酯与内标峰面积比3 针平均值的相对偏差的绝对值均不超过1.63%,表明对照品溶液室温下放置8 h稳定。
3.8 耐用性考察
试验中分别考察了进样口温度变化± 20 ℃、进样口压力变化± 50 kPa、进样时间变化(0.3、0.5、0.7 min)、以及萃取静置时间(0~30 min)对同一甲磺酸样本含量测定结果的影响。结果表明,各项因素下各待测物峰面积的RSD 均不超过3.22%,方法具有较好的耐用性。
3.9 样本测定
取各甲磺酸样本按“2.2.4”项下方法制备供试品溶液,每个样本平行制备两份,按“2.2.3”项下方法制备混合对照品溶液,上述溶液均按“2.1”项下条件GC-MS分析,内标法计算供试品中MMS、EMS、IMS的含量,结果列于表4 Lab 1对应栏目;复核单位验证测定结果列于表4 Lab 2对应栏目。两个实验室对相同甲磺酸供试品中甲磺酸烷基酯的测定结果的相对偏差不超过4.42%,表明所建方法稳定性好、通用性强。
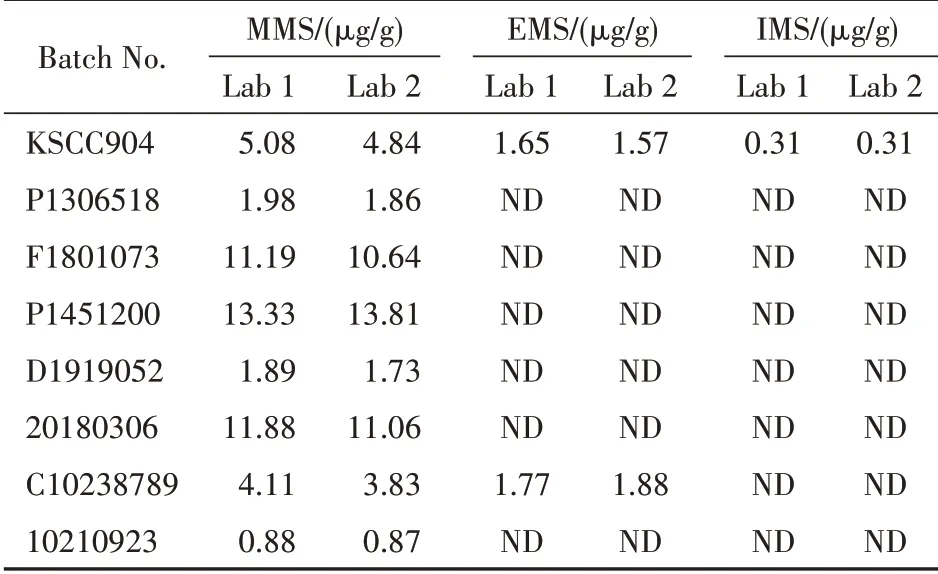
Table 4 Result of content of three alkyl methanesulfonates in methanesulfonic acid samples
4 讨 论
4.1 溶剂和萃取剂选择
采用液液萃取预分离供试品中待测物与主成分,对于毛细管气相色谱法定量测定微量杂质十分重要。甲磺酸易溶于水或醇等溶剂,甲磺酸烷基酯则在弱极性有机溶剂中溶解更好。有文献报道[16]以正己烷为溶剂测定甲磺酸甲酯和甲磺酸乙酯,待测物峰形严重拖尾;Wollein等[17]用正己烷提取甲磺酸溴隐亭中甲磺酸烷基酯时回收率偏低,推测是因正己烷为非极性溶剂,不能有效提取水溶液中甲磺酸烷基酯。二氯甲烷微溶于水,与酯类互溶,沸点适宜,在本试验中被用作甲磺酸烷基酯杂质的萃取剂,水用作甲磺酸供试品的溶剂。
4.2 质谱定量离子及定量方式确定
本试验采用质谱选择性离子监测模式定量测定各待测物。其中,甲磺酸甲酯、甲磺酸乙酯、内标甲磺酸正丁酯分别采用丰度最高的m/z80、m/z79、m/z56离子作为定量离子;而甲磺酸异丙酯,因丰度最高m/z43 离子在检测过程中极易受系统及溶剂等因素干扰[18],影响测定的专属性,定量离子采用丰度略低、但质荷较大的m/z123 离子。为进一步提高检测灵敏度,试验时采用了分时段检测单个离子序列。
4.3 进样方式的优化
甲磺酸中甲磺酸烷基酯的检测属于痕量分析范畴。为提高检测的灵敏度,本试验考察了不分流进样与脉冲不分流进样两种方式的影响,结果见图2。可见,不分流进样测得的色谱图(图2-A)中,各待测物响应偏小且峰的对称性差;采用脉冲不分流进样,当压力在200~300 kPa 范围内时,色谱峰位恒定,各色谱峰峰形对称,甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯对应峰的峰面积分别增加了约134%、77%、140%(图2-B)。这是由于进样口压力增加,各待测物在短时间内快速集中进入色谱柱,谱带展宽小,峰形尖锐且对称度提高。此外,进样口停留时间减少,也降低了待测物质发生热降解的可能。
4.4 色谱柱的比较及采用
采用质谱选择性离子监测时,色谱分离对于甲磺酸中甲磺酸烷基酯的准确测定十分关键。试验初期,参考欧洲药典,采用100%甲基聚硅氧烷为固定液的非极性毛细管柱DB-1(15 m×0.25 mm,1 µm),进行分离分析。通过调整起始柱温、升温速率、流速等色谱条件,发现甲磺酸中待测物与相邻杂质的色谱峰之间很难实现基线分离(图3-A);改用5%苯基-95%甲基聚硅氧烷为固定液的毛细管柱Rxi-5ms(30 m × 0.25 mm,0.25 µm),并尝试优化色谱条件,结果(图3-B)杂质对甲磺酸乙酯色谱峰仍存在影响;当采用HP-1MS(30 m × 0.32 mm,1 µm),并优化色谱条件后,甲磺酸供试品色谱图(图3-C)显示,各待测物峰形尖锐对称,各成分间分离度良好。故最终采用HP-1MS(30 m×0.32 mm,1µm)柱。
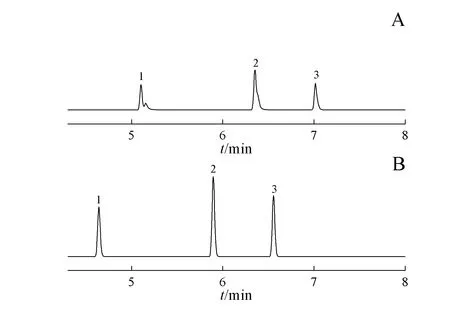
Figure 2 GC-MS chromatograms of plitless injection(A) and pulsed splitless injection under 250 kPa(B)

Figure 3 GC-MS chromatograms of test solution(Innochem,Batch No. KSCC904)in DB-1(A),Rxi-5ms(B)and HP-1MS(C)
4.5 对照品溶液配制方式的改变
欧洲药典以对照品的二氯甲烷溶液作为参照,计算供试品中甲磺酸烷基酯的含量。据此试验,结果均显示:甲磺酸甲酯存在回收率偏低现象(约75%)。考虑到供试品溶液测定前经历了二氯甲烷萃取-无水硫酸钠干燥过程,而对照溶液并无此操作的情况,本研究考察了对照品溶液制备时经萃取-干燥处理与不进行处理对测定结果的影响,结果见表5。
显然,改变对照品溶液配制方式,采用与供试品溶液一样的萃取-干燥处理,可解决应用欧洲药典方法测定甲磺酸烷基酯时结果偏低的问题。
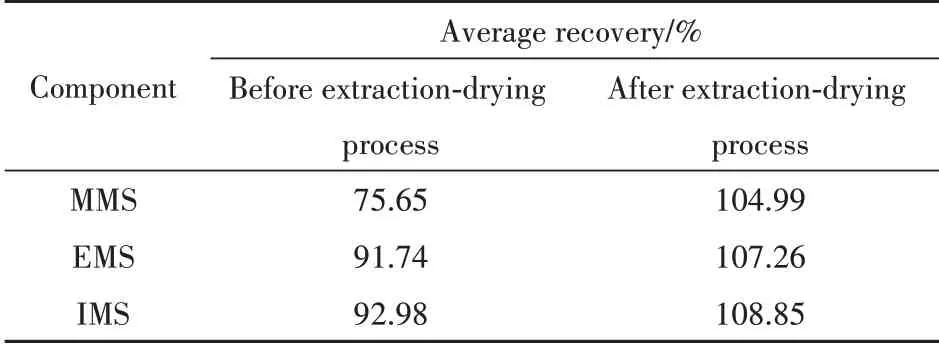
Table 5 Comparison of average recovery before and after extractiondrying process of three alkyl methanesulfonates(n=9)
4.6 定量下限的确定
现行版《中华人民共和国药典》要求分析方法的定量限须满足信噪比不小于10∶1,且准确度、精密度符合要求。为此,本研究考察了甲磺酸烷基酯信噪比约10∶1 时对应的甲磺酸烷基酯质量浓度,约为14.8 ng/mL。进一步平行制备含此浓度的供试品标准添加溶液,考察方法对甲磺酸甲酯、甲磺酸乙酯、甲磺酸异丙酯测定的准确度及精密度。结果发现,精密度均符合要求[8],但甲磺酸异丙酯测得浓度与标准添加浓度存在的相对偏差达39.11%,说明方法对14.8 ng/mL 质量浓度的检测达不到定量要求。提高验证用甲磺酸烷基酯浓度,确定方法的定量限对应质量浓度为37 ng/mL,此时各待测物测定的相对偏差均小于20%。
4.7 标准曲线法和单点校正法的选择
药物中杂质的定量可采用内标标准曲线法或内标单点校正法,本研究对两种方法所得结果的准确度进行了比较。
表6 分别列出了标准曲线法及单点校正法测得的甲磺酸烷基酯低、中、高浓度、理论浓度以及相对偏差。

Table 6 Comparison of standard curve method and single-point calibration method in predicted concentration of three alkyl methanesulfonates
可见,在定量限浓度附近,两种方法的测得值与理论值的相对偏差均小于18%,中、高浓度水平时相对偏差均小于8%,符合现行版《中华人民共和国药典》对分析方法准确度的要求。鉴于测定的便捷性,内标单点校正定量最终被用于本研究建立的分析方法。
5 结 论
本研究建立的液-液萃取结合GC-MS 法具有简便、灵敏、专属、稳定且通用性好的特点,已成功用于甲磺酸中微量甲磺酸甲酯、甲磺酸乙酯和甲磺酸异丙酯的含量测定。
猜你喜欢
石油炼制与化工(2022年6期)2022-06-21
化工设计通讯(2022年5期)2022-05-25
中国典型病例大全(2022年9期)2022-04-19
中国医院用药评价与分析(2022年1期)2022-02-21
中国油脂(2022年1期)2022-02-12
中国畜禽种业(2021年9期)2021-09-22
健康必读·下旬刊(2019年5期)2019-06-03
科技视界(2016年19期)2017-05-18
润滑油(2015年4期)2015-11-20
车用发动机(2015年3期)2015-03-20
