
良性家族性婴儿癫痫的临床特征及遗传学研究
2020-09-12 13:12:02渠蕊戴园园李蕊
临床神经病学杂志 2020年4期
渠蕊,戴园园,李蕊
1岁前起病的良性家族性癫痫可划分为3个癫痫综合征,包括良性家族性新生儿癫痫(BFNE)、良性家族性新生儿-婴儿癫痫(BFNIE)和良性家族性婴儿癫痫(BFIE)。这三种综合征临床表现相似,主要根据起病年龄进行划分。BFIE多于出生3~12个月起病,出生4~7个月为高峰起病年龄,2岁前缓解,预后良好[1]。2012年Heron等[2]首先在14个BFIE家系中发现了PRRT2致病突变,并由此提出PRRT2是BFIE主要致病基因。Zara等[3]研究报道,少数BFIE家系可携带KCNQ2、KCNQ3和SCN2A基因突变。2017年国内文献报道,PRRT2是BFIE的主要致病基因,GABRA6可能是BFIE新的致病基因[4]。本研究对10例BFIE患儿的临床资料进行总结,并采用癫痫基因二代测序检查可疑的致病性突变基因,并对比国内外已报道的BFIE相关致病基因,探讨BFIE的临床特征及致病基因谱。
1 临床资料
1.1 一般资料 系2018年1月至2019年1月徐州医科大学附属医院儿科收治的BFIE患儿10例。BFIE均符合以下诊断标准[1]:(1)起病年龄3~12个月;(2)有良性婴儿癫痫家族史;(3)起病前后智力运动发育正常;(4)表现为局灶性发作或局灶性发作继发全面性发作,可呈丛集性发作(即24 h内发作≥2次);(5)EEG背景正常,发作期EEG为局灶痫样放电,发作间期无典型癫痫样放电;(6)头颅影像学检查无异常;(7)排除低血钙、低血糖等代谢紊乱导致的惊厥。其中男5例,女5例;起病年龄2~14月龄,中位年龄为5月龄;<6月龄起病8例,6~12月龄1例,>12月龄1例。1例出生时有呼吸困难,吸氧后恢复,智力运动发育正常;其余9例患儿出生史无异常,智力运动发育正常。患儿一般临床资料见表1。本研究经徐州医科大学附属医院医学伦理委员会批准,所有患儿监护人均签署知情同意书。

表1 BFIE患儿一般临床资料例序性别起病年龄末次随访年龄发作类型丛集性EEG头颅MRI控制发作药物智力运动发育1女4月15d10月20d部分性发作是正常正常左乙拉西坦正常2男4月7d1岁4月部分继发全面性发作否中央、顶区棘波、棘慢波正常左乙拉西坦正常3女3月5d1岁2月部分性发作是中央、顶区棘波、棘慢波正常左乙拉西坦正常4男6月10d1岁3月部分性发作是正常正常丙戊酸钠正常5女4月15d1岁5月部分性发作否正常正常无正常6男5月2d1岁4月部分性发作否额、中央区棘波、棘慢波正常奥卡西平正常7女3月10d1岁7月部分性发作是正常正常奥卡西平正常8女1岁2月2岁1月部分继发全面性发作否额、中央区棘波、棘慢波正常奥卡西平正常9男7月12d1岁5月部分性发作是额、中央区棘波、棘慢波正常左乙拉西坦正常10男2月16d1岁3月部分性发作是后头部棘波、棘慢波正常左乙拉西坦;苯巴比妥正常
1.2 临床表现
1.2.1 癫痫发作表现 10例患儿均为部分性发作或部分性发作继发全面性发作。发作累及眼、口角、肢体,清醒及睡眠中均有发作,以清醒期发作为主。5例表现为眼凝视、握拳、四肢僵硬或抖动、口唇发紫或面色发绀;3例表现为眼斜视或凝视,动作停止,反应减慢、口唇发紫、呼之不应;2例表现为一手攥拳伴同侧肢体阵发性抖动,渐波及全身,双手攥拳,双上肢屈曲,双下肢伸直伴全身抖动。本组患儿每次发作持续数秒至3 min,无癫痫持续状态。6例患儿发作呈丛集性,1 d≥2次,但发作间期反应良好。1例患儿发作稀少,半年内间断发作2次。
1.2.2 发作伴随症状 2例患儿以发热抽搐起病,发热为低热或高热,之后有间断无热抽搐;其余8例患儿发作期间无伴随症状。
1.2.3 发作性疾病家族史 9例患儿父母有婴儿期发作性疾病史,包括婴儿期起病的无热抽搐史8例,均未治疗,抽搐症状自行消失。
1.3 辅助检查 10例患儿头颅MRI检查均未见异常。10例患儿行24 h视频EEG检查,4例发作间期EEG未见异常;6例发作间期可见散在低-中波幅棘波、棘慢波,分布在额、中央区3例,中央、顶区2例,中央、顶、枕、后颞区1例。1例患儿监测到起源于枕、后颞区的发作期EEG。所有患儿血液检查以及尿代谢检查未见异常。
1.4 基因突变筛查 见表2。留取所有患儿及其父母的静脉血,提取外周血DNA行癫痫基因包靶向二代测序检查。10例患儿均发现基因突变。9例来自父源或母源,其父母一方携带相同致病基因,为家族遗传性突变;1例为新生突变。9例家族遗传性突变中有6例为PRRT2基因突变,包括4例错义突变(c.364C>T、c.986T>C、c.643C>A、c.439G>C)和2例移码突变(c.641delC、c.641dupC),2例SCN2A基因错义突变[c.755(exon7)T>C、c.-18A>C],和1例KCNQ2错义基因(c.241C>T)。除PRRT2基因突变(c.439G>C)已报道外,余8例为未报道的新突变。1例在KCNQ2基因发现的c.2315delC杂合核苷酸变异为移码变异,经验证为BFIE相关的致病基因杂合突变,其父母该位点未发现异常,为未报道的新生突变。综上所述,10例患儿中,除1例PRRT2基因突变位点(c.439G>C)已报道,余9例为未报道的新突变。Sanger测序图见图1、图2。
1.5 治疗及随访 5例采用左乙拉西坦治疗,4例未再发作,1例提高左乙拉西坦剂量后仍有发作,添加苯巴比妥联合治疗后发作控制。3例采用奥卡西平治疗,无发作。1例采用丙戊酸钠治疗后无发作。1例因起病初期发作后一直无发作,故未加抗癫痫药。10例患儿经门诊或电话随访(末次随访年龄10~25月龄,中位年龄16月龄)6~14个月发现,9例予抗癫痫药物治疗的患儿服药后发作易控制,发作停止年龄4~16月龄;1例未用抗癫痫药患儿随访期间未再发作。
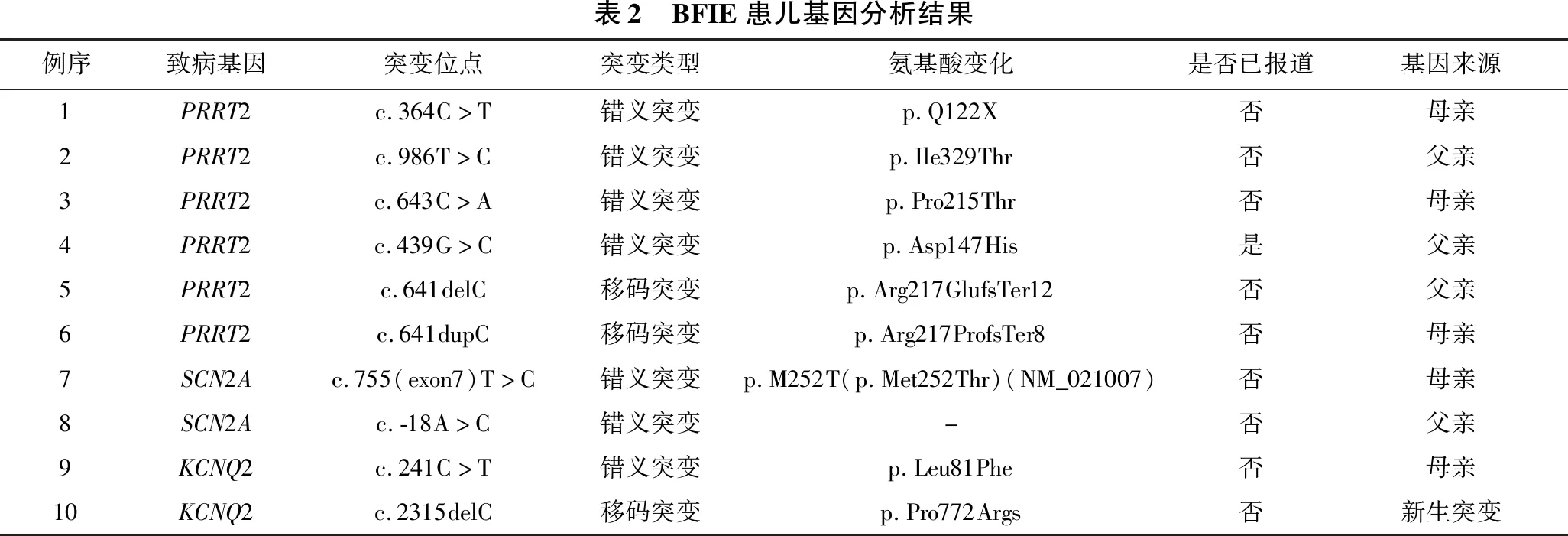
表2 BFIE患儿基因分析结果例序致病基因突变位点突变类型氨基酸变化是否已报道基因来源1PRRT2c.364C>T错义突变p.Q122X否母亲2PRRT2c.986T>C错义突变p.Ile329Thr否父亲3PRRT2c.643C>A错义突变p.Pro215Thr否母亲4PRRT2c.439G>C错义突变p.Asp147His是父亲5PRRT2c.641delC移码突变p.Arg217GlufsTer12否父亲6PRRT2c.641dupC移码突变p.Arg217ProfsTer8否母亲7SCN2Ac.755(exon7)T>C错义突变p.M252T(p.Met252Thr)(NM_021007)否母亲8SCN2Ac.-18A>C错义突变-否父亲9KCNQ2c.241C>T错义突变p.Leu81Phe否母亲10KCNQ2c.2315delC移码突变p.Pro772Args否新生突变
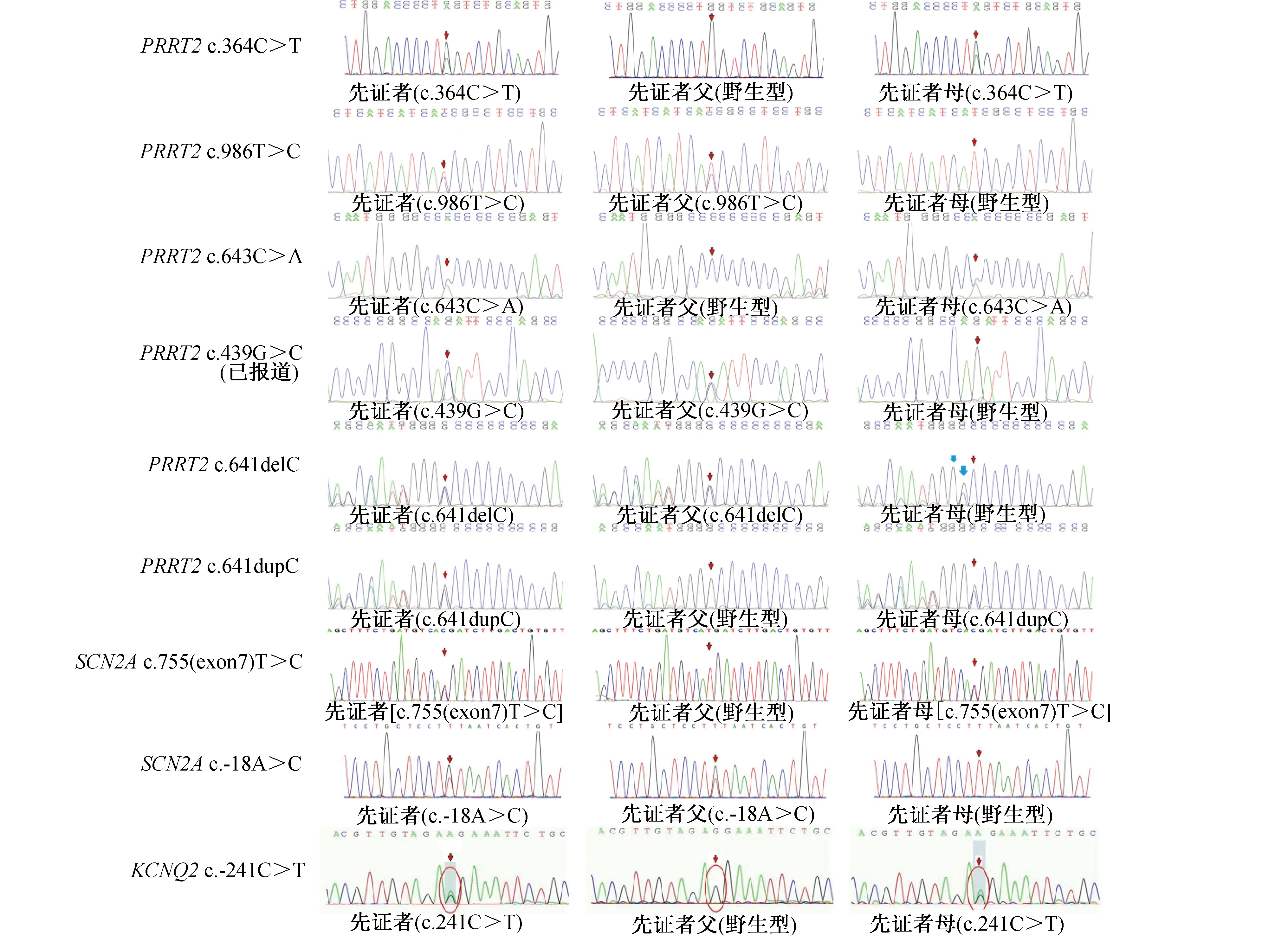
图1 9例家族遗传性先证者及其父母的Sanger 测序图 先证者携带BFIE相关致病基因杂合突变,其父亲或母亲在该基因位点携带相同杂合突变,箭头所指位置或红线圈出部分为突变位点
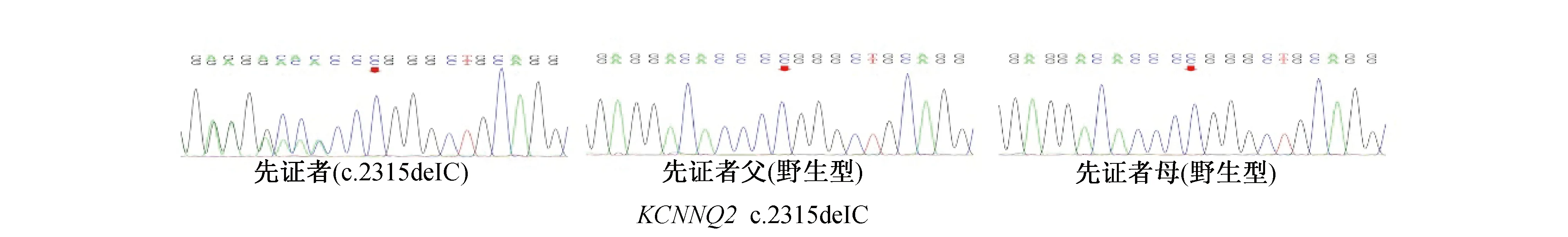
图2 1例新生突变先证者及其父母的Sanger测序图 KCNQ2基因移码突变(c.2315delC)为未报道的新生突变,其父母该位点未见相应突变,箭头所指位置为突变位点
2 讨 论
癫痫是脑部神经元异常放电引起的神经系统疾病,其特征是反复和不可预测的痫性发作[5-7]。BFIE属于良性局灶性癫痫综合征,是1岁内起病的常见癫痫综合征之一。本组病例中5例患儿以左乙拉西坦为首选药物,1例仍有发作。在癫痫患者用药中,症状控制不好时,首选在原用药基础上增加推荐用药剂量的50%,再进一步考虑替换药物或者添加治疗,尽量避免医源性耐药性癫痫的发生[8]。故该例患儿治疗上首选提高左乙拉西坦剂量,后仍有发作,联合用苯巴比妥后很快发作控制。本组3例首选奥卡西平治疗的患儿均为部分性发作,发作间期可见散在低-中波幅棘波、棘慢波,治疗后一直无发作。本组1例首选丙戊酸钠治疗后无发作。但有研究报道,奥卡西平可能加重某些局灶性癫痫患者EEG变化[9],故治疗期间应监测和随访患儿的EEG。1例患儿未用药自行缓解,提示BFIE可为自限性病程。本组4例患儿发作间期EEG未见异常,其余患儿发作间期可见散在低-中波幅棘波、棘慢波(分布在额、中央区3例,中央、顶区2例,中央、顶、枕、后颞区1例),1例患儿监测到发作期EEG(起源于枕、后颞区)。本组患儿均在2岁前发作停止,智力运动发育正常,与文献报道一致[2-3],提示抗癫痫药物可适当缩短这种类型癫痫的病程,一般持续1~2年无发作,患儿可在2~3岁停用抗癫痫药。
BFIE遗传方式为常染色体显性遗传,呈遗传异质性,其致病基因先后定位在3个区间,分别为19q12-q13.1、16p12-q12及2q24。BFIE家系基因突变检出率高,致病基因包括PRRT2、SCN2A和KCNQ2基因[4]。
PRRT2基因位于16号染色体,全长3 794 bp,包含4个外显子,编码340个氨基酸,主要在大脑皮质、基底节和小脑呈高表达。PRRT2基因突变导致癫痫和运动障碍,是发作性运动源性运动障碍、BFIE和婴儿惊厥伴阵发性舞蹈手足徐动症的共同致病基因,也是少数散发性良性婴儿癫痫的致病基因[10]。PRRT2基因相关发作性疾病以自限性婴儿期癫痫、发作性运动诱发的运动障碍为临床表现[11]。目前研究认为,PRRT2基因定位为轴突,并与谷氨酸能突触相关,与突触相关膜蛋白25相互联系,影响钙离子触发的神经递质释放;另外PRRT2基因还影响突触囊泡及电压门控离子通道,突变可能导致突触囊泡释放改变和神经元兴奋性增加,从而引起上述疾病[2]。BFIE家系中PRRT2基因突变最常见。国外文献报道该基因突变阳性家系比率为69%~86%[2-3,12-13];国内文献报道PRRT2基因突变家系占BFIE家系的60.6%[4],稍低于国外。PRRT2基因突变导致的良性婴儿癫痫多数为杂合突变,基因突变多位于蛋白编码区域,主要在第2、3外显子区域,极少数位于内含子区域或剪接位点区域,少数为PRRT2基因整体杂合缺失[14]。2019年李鑫鑫等[15]报道了PRRT2基因主要编码区的2、3号外显子缺失的散发BFIE,推测临床表型及预后与基因突变位点及片段长短无明显相关性。PRRT2基因热点突变为c.649_650insC和c.649delC,其中c.649_650insC最常见[16]。本研究共发现6种PRRT2基因突变,其中4个为错义突变,2个为移码突变,无热点突变。除1例(c.439G>C)为已报道的PRRT2基因突变,5例(c.364C>T、c.986T>C、c.643C>A、c.641delC、c.641dupC)为未报道新突变,扩展了PRRT2基因突变谱。
SCN2A基因位于染色体2q24.3,包含26个外显子,编码2 005个氨基酸。SCN2A基因编码电压门控钠离子通道α2亚基(Nav1.2)。SCN2A基因突变具有表型异质性,该基因是多种癫痫综合征的致病基因,包括预后较好的癫痫如BFNE、BFIE[3,17],以及发育性癫痫性脑病如大田原综合征、婴儿痉挛症、Lennox-Gastaut综合征和不能分类的早发癫痫性脑病等[17]。SCN2A基因突变可为遗传性突变或新生突变。文献报道,遗传性突变常见于BFNE、BFNIE、BFIE和全面性癫痫伴热性惊厥附加症家系,大多预后良好[3];而新生突变多见于癫痫性脑病散发患者,预后不良[18]。本研究2例患儿携带的SCN2A基因突变,均为家族遗传性突变,1例来自父源,另1例来自母源,其父母一方携带相同的致病基因。2例突变为不同的错义突变,无热点突变。2例SCN2A基因突变[c.755(exon7)T>C、c.-18A>C]均为未报道新突变,扩展了SCN2A基因突变谱。SCN2A基因相关的良性(家族性)新生儿/婴儿惊厥患儿中相对有效的药物包括苯妥英钠、奥卡西平、唑尼沙胺、氯巴占[18-22]。治疗中可能出现药物抵抗,包苯巴比妥、托吡酯、丙戊酸钠、左乙拉西坦,部分患儿需接受2个以上(平均4.3个)的抗癫痫药物治疗[18]。本研究2例携带SCN2A基因突变的患儿使用奥卡西平治疗有效,提示SCN2A基因突变导致的癫痫可能对奥卡西平反应好。
KCNQ2位于染色体20q13.3,包含17个外显子(3 251个碱基对)。KCNQ2主要在神经元胞体中表达,编码蛋白为电压门控钾离子通道蛋白亚基Kv7.2。KCNQ2基因与另一个电压门控钾离子通道基因KCNQ3共同编码形成电压门控钾离子通道 M通道。KCNQ2基因遗传方式为常染色体显性遗传。KCNQ2基因突变和KCNQ3基因突变主要与BFNE相关,也有少数报道与BFNIE和BFIE相关[3]。KCNQ2基因相关良性癫痫大多对苯巴比妥和苯妥英钠反应好,少数可能需要使用其他抗癫痫药物才能控制发作[2]。本研究发现2例KCNQ2基因突变,包括1个错义突变(c.241C>T)和1个移码突变(c.2315delC)。携带KCNQ2(c.241C>T)基因突变的患儿与母亲具有相同位点的KCNQ2错义突变,为遗传性突变,首选左乙拉西坦治疗,一直无发作。KCNQ2(c.2315delC)基因突变患儿的父母在该位点未发现异常,为新生突变,采用左乙拉西坦治疗仍有发作,经联合苯巴比妥后很快发作控制。本研究2例患儿携带的KCNQ2基因突变,均为未报道的新突变。2例患儿目前智力、运动发育均正常。
BFIE患者癫痫发作对抗癫痫药物敏感,且多呈自限性,预后良好。PRRT2、SCN2A和KCNQ2基因是BFIE的致病基因,PRRT2基因突变阳性率最高。对于怀疑BFIE的患儿可以考虑早期进行基因突变筛查,有利于明确诊断、选择治疗方案、指导抗癫痫药物的使用疗程及判断预后,同时对指导家庭遗传咨询具有重要作用。
猜你喜欢
广东药科大学学报(2023年5期)2023-12-30 00:08:39
临床儿科杂志(2023年8期)2023-09-29 09:38:52
时代报告·奔流(2022年1期)2022-04-29 04:10:56
昆明医科大学学报(2022年3期)2022-04-19 13:59:42
昆明医科大学学报(2021年10期)2021-12-02 03:24:38
中华养生保健(2020年1期)2020-11-16 00:47:46
作文周刊·小学四年级版(2018年40期)2018-04-09 08:20:04
中国当代医药(2015年30期)2015-03-01 02:08:09
浙江医学(2014年17期)2014-04-13 10:13:16
海南医学(2013年3期)2013-04-08 10:46:12
