
13号染色体q22.1q32.3间隙缺失致多发畸形1例并文献复习
2020-08-28 08:39:36宋亚蓝赵芷若张云峰刘秀敏
中国实验诊断学 2020年8期
宋亚蓝,黎 萍,邢 杰,赵芷若,张云峰,刘秀敏
(1.吉林大学第二医院 发育儿科,吉林 长春130041;2.吉林大学第二医院 检验科,吉林 长春130041)
13号染色体长臂片段间隙缺失是非常罕见的染色体异常,多源自新发的突变,其表型主要取决于缺失的部位及片段大小[1]。本文报道回顾分析1例生长发育落后、特殊面容伴多发畸形,经外周血染色体核型分析示13q22-32 缺失患儿的临床表型及其基因型的相关性,并与既往报道的病例进行对比及分析。
1 临床资料
患儿,男,汉族,9 个月,因生长发育落后及多发畸形就诊。患儿系 G1P1,足月顺产儿,出生体重 2 100 g(<-2SD),身长 40 cm(<-3SD),为小于胎龄儿,孕期无宫内窘迫,生后无窒息史。生后母乳喂养,生后无住院病史、无手术史。4个月能竖头,6个月会翻身,不会独坐,不会爬,不会扶物站立。
家族史:父、母生育年龄均为 23 岁,否认近亲结婚。其母既往健康,孕期无异常接触史及用药史。家族中无发育落后、骨骼发育异常等病史。
体格检查:神清,精神反应尚可,浅表淋巴结无肿大,身高65 cm(<-3SD),体重6.1 kg(<-3SD),头围 39 cm(<-3SD),眼距宽,眼裂小,追视、追听良好,上腭高、窄,双耳位低,左手拇指缺如,手指及足趾短,掌纹浅(图 1、2),心肺听诊无阳性体征,腹部触诊未触及异常包块,阴茎小(长约1.0 cm),双侧隐睾;神经系统检查未见异常。心脏彩超检查未见异常,头部核磁共振未见明显异常。

图1 本病例的面容

图2 本病例的双手
Gesell 发育评估:运动能 DQ大运动 65(相当于 6 个月)、精细运动 70(相当于 6.5 个月)、应物能 DQ76(相当于 7 个月)、言语能 DQ76(相当于 7 个月)、应人能 DQ76(相当于 7 个月)提示全面发育迟缓。
经过患儿父母知情同意并签署知情同意书后,经过吉林大学第二医院医学伦理委员会批准,抽取患儿及其父母静脉血样进行细胞遗传学和分子遗传学分析。外周血染色体核型分析(550 带,G 带)抽取外周新鲜抗凝血1 ml,加入10 ml含20%小牛血清的培养基中,然后置于5%CO2培养箱37℃培养72 h,收获前1 h加入终浓度为8×10-5g/L的秋水仙素。常规方法收获淋巴细胞、悬浮滴片、烘干、G显带。镜下分析50个染色体中期分裂相,镜下拍摄配对分析5个中期核型,核型描述参考《人类细胞遗传学国际命名体制》(ISCN2009)。
采取患儿2 ml静脉血,应用QIAGEN试剂盒提取基因组DNA。应用美国Affymetrix公司生产的CytoScanHD(195万CNV探针+75万SNP探针)芯片对样本进行检测。对所得的原始数据应用Affymetrix Chromosome Analysis Suite Software进行分析。收集文献报道,并参照国际基因组CNV多态性数据库DGV、UCSC Oenome Browser(https://genome.ucsc.edu/)、DECIPHER以及ISCA数据库及相关文献判读CNV缺失区域,结合OMIM基因分析,分析可能导致多发畸形、全面发育迟缓的基因。
常规染色体G显带结果显示患儿染色体核型为46,XY,del(13)(q22q32),如图3所示;父母核型均未见明显异常。染色体微阵列比较基因组杂交(aCGH)分析 患儿的13号染色体在13q22.1q32.3区域有28.32Mb 的缺失(arr[hg19]13q22q32(72,800,306-101,119,087)×1)。通过查询DECIPHER和OMIM数据库,发现该区域含有15个编码蛋白的基因:PIBF1、TBC1D4、CLN5、EDNRB、SPRY2、SLITRK1、SLITRK6、MIR17HG、GPC6、TGDS、CLDN10、DNAJC3、ZIC2、PCCA、NALCN,其中缺失可导致显性致病的基因有NALCN、ZIC2、SPRY2、EDNRB和MIR17HG,易感基因有SLITRK1。随访至15个月,患儿会爬,不会独站、独走,会有意识喊“爸爸、妈妈”。

图3 本病例的染色体核型:46,XY,del(13)(q22q32)
总之,本例13q22.1-q32.3 微缺失所致全面发育迟缓、腭弓高、耳位低、双侧隐睾、小阴茎和手足畸形,通过分析发现MIR17HG 基因杂合缺失是导致该表型的关键基因。本例也表明aCGH染色体微阵列是临床上发育迟缓、多发畸形患儿分子诊断的重要工具。
2 讨论
该患儿为足月小于胎龄儿,眼间距宽,眼裂小,少指,隐睾、小阴茎,生后体格、神经运动发育落后,首先需要注意常染色体病,因而检测其外周血染色体核型,发现该患儿存在 13 号染色体q22-q32缺失;aCGH分析进一步验证该患儿13q22.1q32.3区域有28.32 Mb的微缺失。该区域包含多个致病基因,其中可显性致病的基因有NALCN、ZIC2和MIR17HG,易感基因有SPRY2、EDNRB、SLITRK1。NALCN 基因杂合缺失可导致四肢和面部的先天性挛缩,肌张力低下和发育迟缓综合征(Congenital Contractures Of The Limbs And Face,Hypotonia,And Developmental Delay;CLIFAHDD)[2]。ZIC2 基因缺陷可导致前脑无裂畸形5型(Holoprosencephaly 5;HPE5)[3],前脑无裂畸形是人类大脑最常见的结构异常。位于 13q31.3 的 MIR17HG 基因缺陷可导致Feingold 综合征2 型,表现为手指和脚趾异常,第二指和第五指短,第五指向内弯曲,拇指发育不全,以及第二和第三脚趾融合,或者第四和第五脚趾的融合,小头畸形,小颌,睑裂小,轻度至中度学习障碍[4];少部分患者还有心脏和肾脏畸形、椎体异常和耳聋[5]。位于 13q31.1 的SPRY2 基因缺失可增加 IgA 肾病易感性[6-9]。位于 13q22.3 的 EDNRB 基因突变可增加先天性巨结肠(Hirschsprung’s disease,HSCR)易感性[10-12]。位于 13q31.1 的 SLITRK1 基因缺失增加图雷特综合症(Tourette syndrome,TS)的易感性,TS是一种以持续的声音和运动抽搐为特征的神经精神发育性疾病[13-17]。结合本例患儿临床资料,MIR17HG 基因的杂合缺失是为该患儿体格、神经运动发育迟缓、面容异常以及手、脚多发畸形的关键致病基因。
本文回顾了13号染色体长臂q22-q32间隙缺失文献,表1列出了目前报道的包括本病例在内的 12例13号染色体q22-32 区域缺失的临床表现,常见的临床特征有全面发育迟缓/智力障碍(9例)、生长发育落后(11例)、肌张力低下(6例)、小头畸形(6例)、宽鼻梁(8例)、耳位低(9例)、手畸形(11例)、足畸形(9例)、高腭弓(6例)、小颌(6例),不常见的临床特征有上门牙突出(4例)、脑积水(2例)、隐睾/小阴茎(5例)、先天性巨结肠(3例)、先天性髋关节发育不良(5例)、癫痫(2例)、中枢神经系统畸形(3例)。
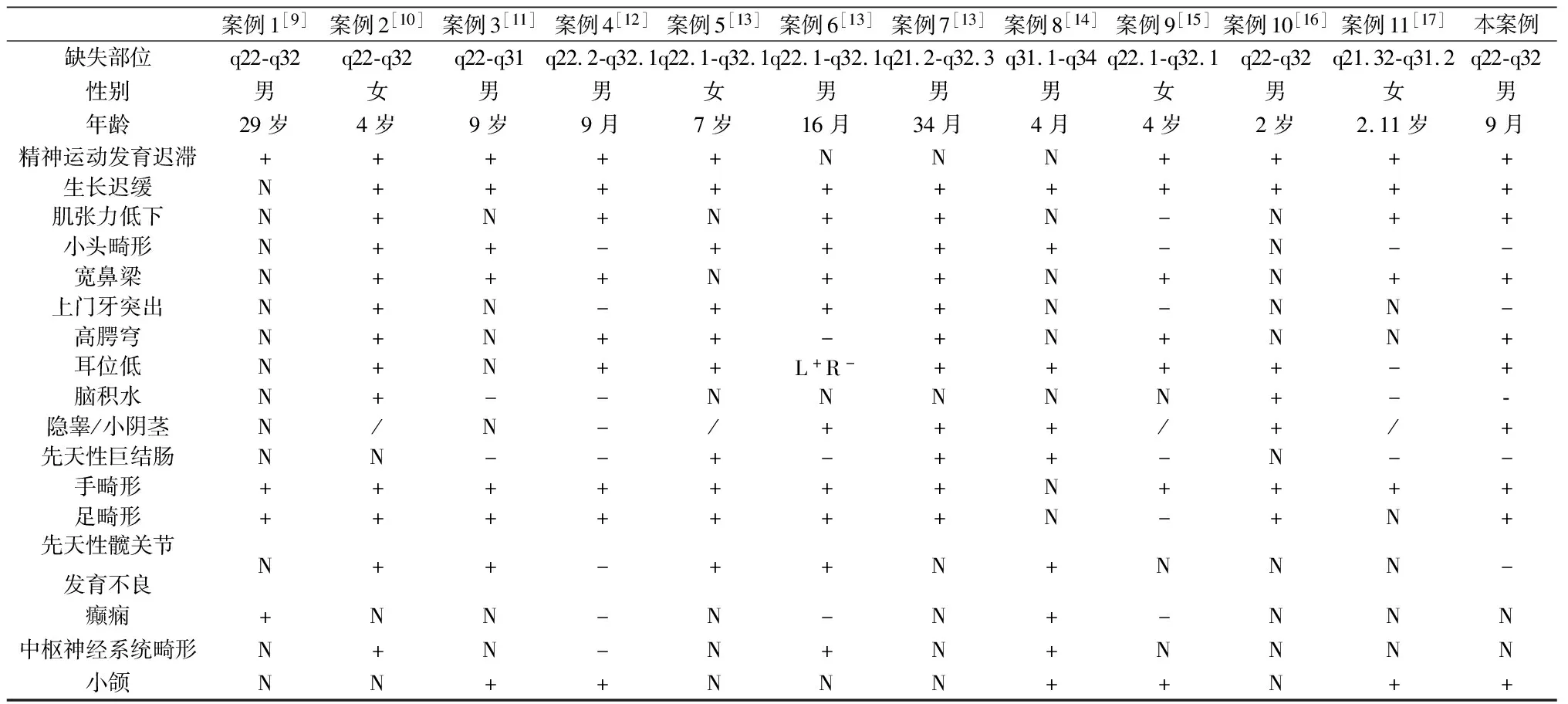
表1 12例 13q22-32 缺失病例的常见临床表型比较
猜你喜欢
保健与生活(2022年7期)2022-04-08 21:33:36
宁夏医学杂志(2020年3期)2021-01-21 08:23:24
科学之谜(2019年3期)2019-03-28 10:29:44
科学之谜(2018年8期)2018-09-29 11:06:46
中国医药指南(2017年3期)2017-11-13 02:58:44
恋爱婚姻家庭·养生版(2016年9期)2016-09-07 11:25:01
哈尔滨医药(2015年2期)2015-12-01 03:57:21
西南医科大学学报(2015年1期)2015-08-22 13:01:50
中央民族大学学报(自然科学版)(2015年2期)2015-06-09 08:45:16
中国当代医药(2015年9期)2015-03-01 02:02:12
