
基于单细胞测序分析三阴性乳腺癌中巨噬细胞标记基因及其功能
2020-08-14 06:08祝佩瑶韩亚东王人颢
医学研究生学报 2020年7期
许 涛,杨 军,韩 咏,祝佩瑶,杨 勇,韩亚东,庄 磊,王人颢,宋 军,张 斌
0 引 言
目前,常规的测序方法主要是对血清或者病理组织进行测序,获得的只是无数细胞混合的DNA样本,而其中单个细胞的独有特性往往被忽视[1]。而单细胞测序技术能够提供每个细胞的RNA表达谱,甚至可以鉴定出组织中既往未被发现和研究的稀有亚群细胞[2]。在高度复杂的肿瘤微环境中,单细胞测序技术则能更加详细地了解其中的细胞信息[2]。
乳腺癌是女性最常见的恶性肿瘤之一,居中国女性癌症发病首位,发病率和死亡率均呈上升趋势[3]。其中三阴性乳腺癌(triple-negative breast cancer, TNBC)是一种异质性疾病,较其他分型的乳腺癌具有发病年龄低,侵袭性强,复发和转移早的特点,且缺乏标准治疗手段,疗效不佳,预后差,严重影响患者的身心健康[4]。为了揭示乳腺癌分子生物学的启动事件,了解其异质性的本质,进而识别可能作为预后生物标志物的异常癌基因,并为乳腺癌患者开发分子靶向治疗提供新的方法思路,于是,单细胞测序技术积极的被用于乳腺癌的研究之中[5]。乳腺癌有不同的类型,其肿瘤微环境亦不同,我们可以查阅到对乳腺癌相关成纤维细胞,上皮细胞的不同亚群图谱的相关文献[6-7],但是,关于TNBC中巨噬细胞的单细胞测序研究尚未见报道。
本研究对TNBC癌组织中超过1000个细胞的表达数据进行单细胞分析,将不同的细胞聚类为不同的亚组,并对其进行注释,得到不同种类的亚群细胞的标记基因。随后,对于巨噬细胞的标记基因,进行基因本体论(Gene Ontology,GO)和京都基因与基因组百科全书(Kyoto Encyclopedia of Genes and Genomes,KEGG)富集分析以及蛋白-蛋白互作(protein-protein interaction,PPI)网络的构建,并最终获得5个基因簇及其中4个基因簇的关键基因。
1 材料与方法
1.1数据来源和质控数据下载自GEO数据库GSE118389数据集中[8]。本数据集从6个TNBC患者的癌组织中提取了1534个独立细胞。测定基因数目>10 000,<50和线粒体基因百分率>5%的细胞。随后,将不同细胞之间变异系数较大的1500个基因挑选出进行后续分析。
1.2聚类分析为减少单细胞自身以及批次效应而导致的基因表达不稳定性,并保留其中大部分的基因表达信息。主成分分析(principal component analysis,PCA)和t分布随机邻接嵌入(t-Distributed Stochastic Neighbor Embedding,t-SNE)聚类。PCA和t-SNE聚类结果是在R语言中使用“BiocManager”、“GSVA”、“GSEABase”包分析获得。
1.3不同亚群细胞标记基因的筛选及注释对于以上分析获得的15个亚群细胞,分别对每个亚群与其他所有亚群的基因表达平均值进行比较,获得每个亚群较高表达的基因集合,即标记基因。随后,在R语言中,通过“SingleR”包,对比既往公认的各细胞标记基因,从而注释了各亚群细胞所对应的细胞类型。
1.4GO功能富集分析和KEGG信号通路富集分析利用DAVID数据库(http://david.ncifcrf.gov/)对巨噬细胞的标记基因进行GO功能富集和KEGG信号通路富集分析[9],了解标记基因可能参与的生物学过程、分子功能、细胞定位及信号通路等。以基因数目>10且P≤0.05为差异有统计学意义。
1.5PPI网络的建立及利用Kaplan-Meier Plotter数据库进行生存分析利用STRING数据库(http://string-db.org/)以及Cytoscape软件联合分析构建巨噬细胞标记基因蛋白互作网络[10]。利用Cytohubba插件依据连接度筛选出关键基因。Kaplan-Meier plotter数据库(http://kmplot.com/analysis/)整合了TCGA、GEO、EGA数据库中乳腺癌等癌症的mRNA与miRNA的表达和预后数据[11]。本研究利用Kaplan-Meier plotter(http://kmplot.com/)数据库进行TNBC患者生存分析,预后评价指标为总生成率(overall survival,OS)。
1.6TIMER数据库分析利用TIMER数据库(https://citrome.shinyapps)对关键基因表达水平与TNBC癌组织中巨噬细胞浸润状态进行相关性分析,得出相关关系散点图[12]。利用Spearman相关系数(cor值)和P值对基因表达与肿瘤免疫浸润相关性进行评价,cor>0为正相关,cor<0为负相关,P≤0.05时认为两者差异具有统计学意义。相关性的强弱与cor的绝对值的大小有关,设定标准:0.00~0.39“弱相关性”、0.40~0.59“中等相关性”、0.6~1.0“强相关性”。
2 结 果
2.1聚类分析结果PCA降维后选取前20的主成分,并进行t-SNE聚类分析,获得了15个聚类的细胞群,不同类的细胞群标记为不同的颜色,见图1。
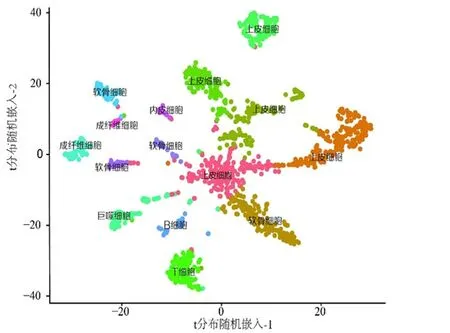
图 1 PCA及t-SNE聚类分析
2.2标记基因的筛选通过比较每一类与其他类别细胞群中基因表达的平均值,其中共找到244个高表达巨噬细胞的标记基因。巨噬细胞部分高表达标记基因有RNASE1、RGS1、CD163、FCGR3A、HLA-DA、HLA-DRB5、HLA-DRB1、HLA-DPA1等。
2.3富集分析结果结果显示:在生物学功能方面,主要富集于嗜中性粒细胞脱颗粒,中性粒细胞激活等;在细胞组分方面,主要富集于分泌颗粒膜,分泌颗粒内腔,细胞质囊腔之中;在分子功能方面,主要富集于酰胺绑定,肽绑定,细胞黏附分子结合等。KEGG富集分析显示,巨噬细胞的标记基因主要富集于肺结核、利什曼病、金黄色葡萄球菌感染等疾病的信号通路中。见表1。
2.4PPI网络及预后分析PPI网络图分析出了主要5个关键模块。并筛选出PPI网络中节点度前4的关键基因,分别为CSF1R、HLA-DQA1、LAPTM5和OLR1。见图2。预后分析表明CSF1R、HLA-DQA1、OLR1基因的高表达时TNBC患者总生存率(OS)高于低表达时(P<0.05)。LAPTM5基因表达对TNBC患者的OS的影响差异无统计学意义 (P>0.05)。见图3。
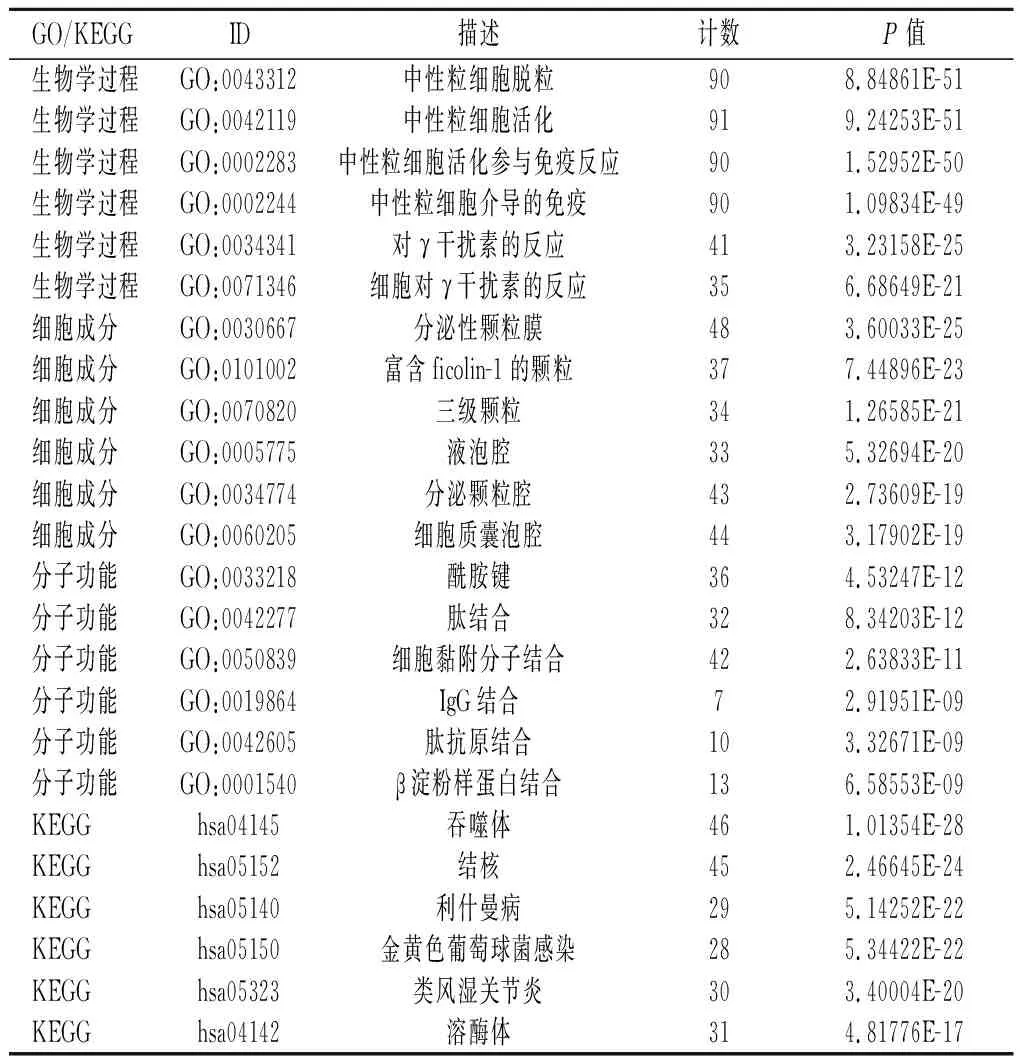
表 1 巨噬细胞GO和KEGG富集分析
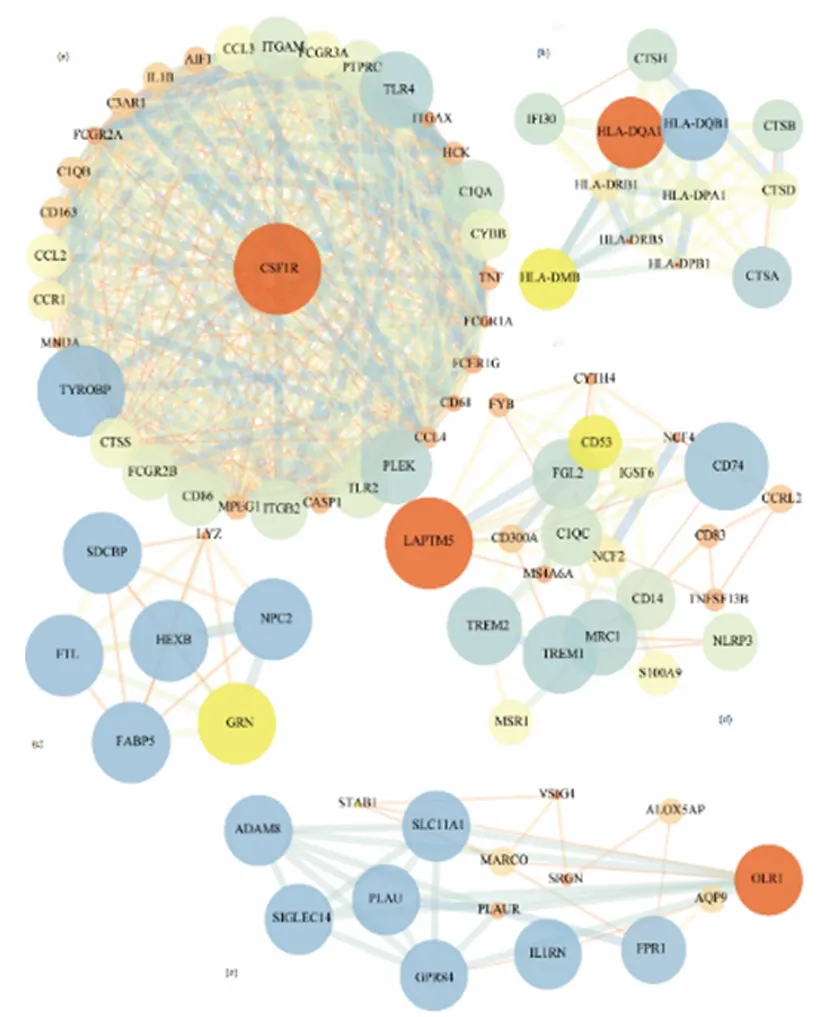
红色标记基因为每个模块的关键基因

a:CSF1R; b:HLA-DQA1; c:LAPTM5; d:OLR1
2.5关键基因表达水平与巨噬细胞浸润情况相关性分析结果CSF1R、HLA-DQA1、LAPTM5、OLR1基因在肿瘤微环境中的表达与肿瘤纯度均具有负相关性(P<0.05),即CSF1R、HLA-DQA1、LAPTM5、OLR1均是主要表达于肿瘤微环境而非肿瘤组织中。CSF1R、LAPTM5、OLR1基因表达水平与巨噬细胞浸润状态成正相关性(P<0.05);而HLA-DQA1基因表达水平与巨噬细胞的浸润状态差异无统计学意义(P>0.05)。在TNBC的肿瘤微环境中,OLR1、CSF1R、LAPTM5基因表达水平越高,巨噬细胞的浸润水平越高。见表2。
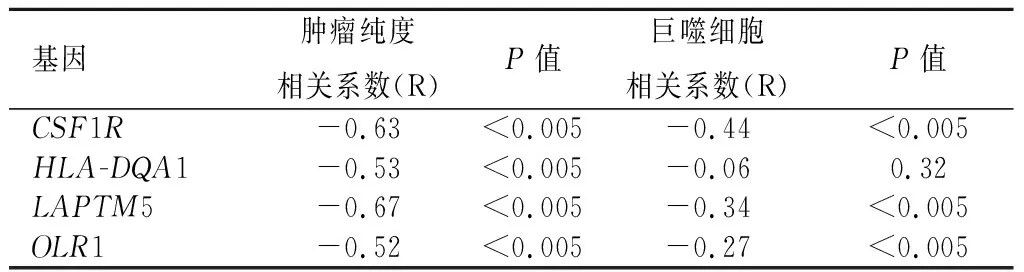
表 2 基因与肿瘤纯度、巨噬细胞相关性分析结果
3 讨 论
巨噬细胞约占肿瘤组织中细胞成分的50%以上[13]。巨噬细胞原本应该消灭肿瘤细胞的,然而,研究发现其却能和肿瘤细胞和谐相处,甚至起到促进肿瘤进展和转移的作用[14]。这是因为肿瘤十分狡猾,通过多种方法骗过了巨噬细胞,甚至“拉巨噬细胞入伙”,反过来帮助肿瘤细胞。例如,肿瘤细胞可以表达CD47,并伪装成正常细胞来诱骗巨噬细胞,同时表达MHC1,减少巨噬细胞表面的LILRB1受体表达,抑制巨噬细胞对肿瘤细胞的吞噬[15]。此外,热休克蛋白27可以导致单核细胞分化为具有免疫耐受表型的巨噬细胞,并进一步诱导T细胞对肿瘤细胞的杀伤作用减弱甚至消失[16]。因为巨噬细胞深入参与到肿瘤的各个过程且肿瘤中巨噬细胞数量庞大,所以巨噬细胞拥有巨大的抗肿瘤潜能,多种巨噬细胞的标记基因可能参与了肿瘤发生,发展,甚至转移的多个生物学过程。通过对巨噬细胞标记物的靶向治疗[17],从而恢复巨噬细胞本身,甚至整个免疫系统的抗肿瘤作用,或许能够对肿瘤细胞的抑制与杀伤起到至关重要的作用。
在本研究中,我们从GEO数据库中获取了数千个TNBC癌组织细胞的表达谱,并过滤了其中表达量较多或者较少的细胞,以及在所有基因表达无明显变化的细胞。由于过滤后的细胞数据仍然很大,所以我们利用PCA和t-SNE分析将数据降维,最终只获得了15个细胞集合,找到了各个细胞集合之间的标记基因,并对各个细胞进行了注释从而知道了细胞的具体种类,得到我们想要研究的巨噬细胞。
值得注意的是,现在肿瘤中的巨噬细胞,主要分为M1、M2型巨噬细胞,肿瘤相关巨噬细胞,其中M1型巨噬细胞起到抑制肿瘤的作用,M2型巨噬细胞和TAMs则具有促进肿瘤进展的作用[18]。通过查阅文献,我们分别找到了不同巨噬细胞的公认标志物,分别为M1型巨噬细胞标志物(IRF5,NOS2,PTGS2);M2型巨噬细胞的标志物(CD163,MS4A4A,VSIG4);TAMs的标志物(CCL2,CD68,IL10),并从我们获得的244个标记基因中寻找以上标志物,发现CD163,MS4A4A,VSIG4,CCL2,CD68均出现在我们的标记基因中,即我们获得的巨噬细胞类型很有可能是M2型巨噬细胞和TAMs[19-22]。
通过应用STRING数据库对244个巨噬细胞标记基因构建PPI网格图,并利用Cytoscape软件处理后获得了4个关键基因,CSF1R、HLA-DQA1、LAPTM5、OLR1。随后,我们查阅文献没有发现既往有关于以上关键基因在TNBC中的预后报道,于是运用Kaplan-Meier Plotter数据库进行生存分析,发现其中CSF1R、HLA-DQA1、OLR1的表达与TNBC患者总生存率存在明显负相关性,表达越高,预后越差。而且,我们使用了TIMER数据库分析了关键基因表达水平与巨噬细胞浸润情况的相关性,发现CSF1R、LAPTM5、OLR1基因表达水平与巨噬细胞浸润状态成正相关性。因此,我们猜测CSF1R、HLA-DQA1、OLR1基因在巨噬细胞中的总体表达作用可能促进TNBC癌组织细胞的增殖、侵袭和迁移,有可能作为判断TNBC预后的因素。
我们查阅文献发现CSF1R被报道可以减少乳腺癌中的巨噬细胞蓄积并减慢乳腺肿瘤的生长速度[23],可能成为TNBC靶向治疗的新靶点[24];LAPTM5可以在巨噬细胞中分泌细胞因子并促进炎症信号通路[25];HLA-DQA1参与了乳腺癌患者帕拉替尼介导的肝损伤[26];ORL1可以被癌基因TBC1D3上调,从而促进人乳腺癌细胞迁移,但在乳腺癌的巨噬细胞中的作用尚未见报导[27]。至于关键基因在巨噬细胞中的表达对TNBC预后的影响这在未来还需要进一步通过实验来分析与研究。
综上所述,本研究通过对TNBC癌组织单细胞数据处理,我们获得了244个巨噬细胞的标记基因,并了解到这些标记基因主要富集的生物学过程,这些标记物未来可能作为攻克肿瘤的重要靶点。获得的4个巨噬细胞关键基因CSF1R、HLA-DQA1、LAPTM5和OLR1,其中CSF1R、HLA-DQA1、OLR1的较高表达提示TNBC患者可以拥有较高的生存率。当然,本研究并不完善,各种标记物的效果仍然需要更多单细胞数据的验证,此外,对于关键基因,我们并未深入研究,需要后续的实验进一步的探索。
猜你喜欢
中华骨与关节外科杂志(2022年1期)2022-08-31
中国医药科学(2022年5期)2022-05-05
中国生殖健康(2020年4期)2021-01-18
科学(2020年4期)2020-11-26
教育界·上旬(2020年8期)2020-06-27
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
江苏农业科学(2017年16期)2017-10-27
中国卫生标准管理(2015年3期)2016-01-14
