苯并噻唑功能化的金属螯合剂的合成及生物活性
2020-07-20 02:06王江淋
无机化学学报 2020年7期
孙 斌 王江淋
(1重庆高校天然药物研究重点实验室,重庆 400067)
(2重庆工商大学环境与资源学院,重庆 400067)
阿尔茨海默病(Alzheimer′s disease,AD)是一种常见的与年龄相关的老年性疾病,截止到2016年,全球约有4 600万人患有这种老年性疾病[1]。迄今为止,还没有真正意义上的治疗AD的方法,其诊断的高准确性来自于对因该病致死患者脑部的尸检。AD最典型的病理特征是神经细胞外的淀粉样斑块(amyloid plaques,AP)和细胞内的神经缠结(neurofibrillary tangles,NFT)[2]。AP 的主要成分是β-淀粉样肽(βamyloid peptide,Aβ)[3]。Aβ的主要异形体是分别由 42 和 40 个氨基酸残基组成的肽 Aβ1~42和Aβ1~40[4-5]。Aβ1~40在大脑中含量较高,但Aβ1~42则具有更大的神经毒性、并且更具聚集倾向。根据Aβ级联假说,Aβ的过度产生与积聚会促进Aβ寡聚物、原纤维以及最终导致神经变性的纤维生成。然而,近期研究显示,可溶性Aβ寡聚物可能更具神经毒性,可能会导致AD患者以及AD动物模型中的触突功能障碍与记忆丧失[6-9]。在这方面,由于缺乏对各种Aβ聚集形态的神经毒性作用的全面了解,20多年来,基于Aβ级联假说研发的治疗AD的药物都遭到失败。
在AD患者大脑淀粉样板块中,发现有高浓度的 铜 (394 µmol·L−1)、锌 (1 055 µmol·L−1)和 铁 (940µmol·L−1),研究表明金属离子能与Aβ相互作用并促使淀粉样斑块的形成[10-13]。这些金属离子不仅加速了Aβ聚集,而且具有氧化−还原活性的金属(如铜、铁)还会导致活性氧(reactive oxygen species,ROS)的生成与氧化应激[14-18]。鉴于Aβ与过渡金属离子的相互作用,一些体外实验研究发现金属螯合剂能降低金属-介导的Aβ聚集、减少ROS的生成、降低神经毒性[19-22]。有几种螯合剂如去铁按(desferrioxamine,DFO)、DP-109、氯碘羟喹(clioquinol,CQ)等(图 1)已被评估在转基因小白鼠试验中取得成功[23-25]。这些螯合剂除螯合金属离子外,还能设计成具有抗氧化性能的自由基清除剂。因此金属螯合剂疗法可能是一种有前途的AD治疗策略。尽管像CQ这样的螯合剂能有效抑制Aβ聚集并改善认知,但是CQ不会与Aβ-金属选择性地相互作用,会出现副作用,这可能会限制其长期的临床应用。现有的研究表明,对Aβ具有强亲和力的配体更能有效地阻止Aβ聚集和抑制Aβ的神经毒性。使用多重同时作用来增强靶点结合亲和力及特异性是生物学中的重要概念且用途广泛。相比较非特异性螯合剂,能识别Aβ的配体显示出了更强的阻止纤维状Aβ形成的能力[26-27]。
众所周知,苯并噻唑经适当修饰后与β-淀粉样斑块有良好的结合性,是某些β-淀粉样蛋白的显像剂的重要组成部分。曲晓刚教授的研究表明,苯并噻唑修饰的螯合剂能有效抑制铜离子诱导的Aβ聚集,并能将聚合的Aβ-Cu加合物中的Cu螯合出来,使聚集的Aβ解聚[28]。因此我们设计合成了2种苯并噻唑修饰的螯合剂(L1、L2,图2),研究了它们抑制金属离子诱导的Aβ聚集,对其控制Aβ-Cu产生H2O2的量以及缓解Aβ聚集产生的神经毒性等生物活性进行研究,并与螯合部位结构相同但没有苯并噻唑功能化的螯合剂(L3)进行了对比。
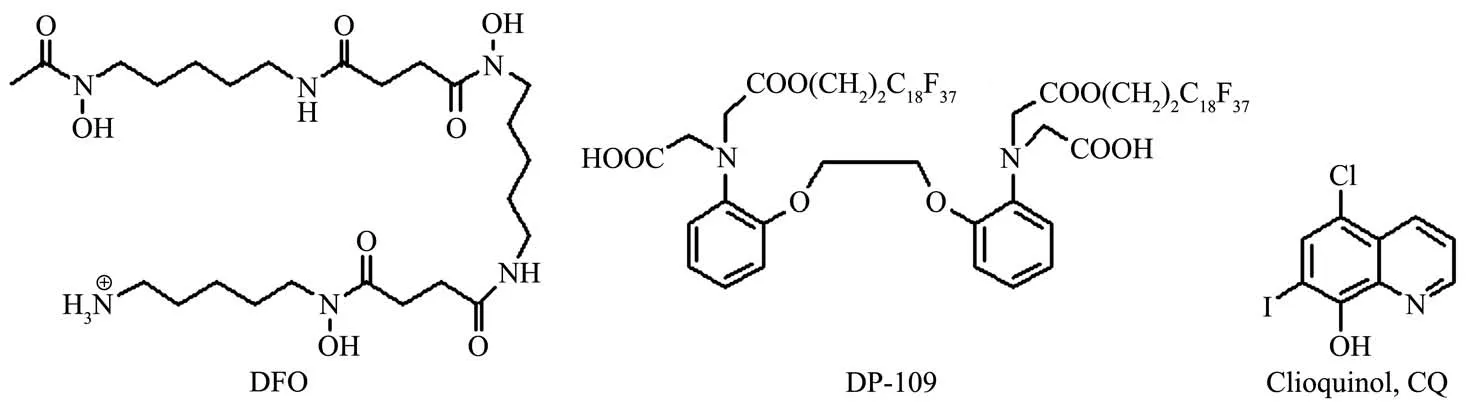
图1 几种重要的治疗AD的金属螯合剂Fig.1 Several important chelators for AD therapy
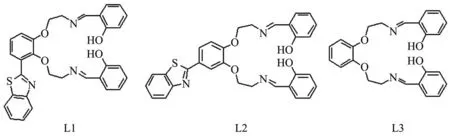
图2 能识别Aβ的金属螯合剂的结构Fig.2 Structure of metal chelators with function of recognizing Aβ
1 实验部分
1.1 仪器与试剂
所有试剂都是从试剂公司购买,如未特别指明,使用前所有试剂都未进行特别处理。螯合剂L3是已知化合物,由本实验室按文献[29]方法自制。反应进程由薄层层析监控,斑点用紫外灯或含有Ce(NH4)2(NO3)6(0.5 g) 和 (NH4)6Mo7O24·4H2O(24.0 g)的6%H2SO4(500 mL)的黄色溶液显色。反应产物的核磁共振氢谱由Varian mercury plus 400 MHz核磁共振仪(TMS为内标)测定,ESI质谱由Q-TOF Ultima API LC-MS液−质联用仪分析确定。
Aβ储备液是将 Aβ1~40(人)(2 mg)溶于氢氧化钠(500 µL,20 mmol·L−1),经超声处理30 s后,用超纯水(1.5 L)稀释,再超声处理 30 s后,用 0.1 mol·L−1的盐酸调pH值到7.4,用0.22µm的滤器(Millipore)过滤,Aβ储备液浓度由BCA蛋白试验测定后置于−20℃下 保 存 。Zn2+和 Cu2+(200 µmol·L−1,以 pH=7.4 的HEPES(2-(4-(2-hydroxyethyl)-1-piperazinyl)ethanesulfonic acid)缓冲液配制)分别用Aldrich原子吸收标准溶液配制。螯合剂L1,L2和L3分别溶于DMSO中配成最终浓度为4 mmol·L−1的溶液。实验所用水均为蒸馏去离子水用超滤膜过滤所得,所用仪器还有TECAN多功能酶标仪(瑞典)。
1.2 合成
螯合剂按Scheme 1合成。
3-(苯并[d]噻唑-2-基)-1,2-苯二酚(3a):2-氨基苯硫酚(1)(2.5 g,20 mmol)与 2,3-二羟基苯甲醛与无水乙醇(40 mL)混合,回流搅拌24 h。冷却后,有大量固体出现,过滤,滤饼用冷乙醇洗涤2次后,用乙酸乙酯重结晶得到3a(3.65 g,75%)。1H NMR(400 MHz,DMSO-d6):δ9.66(s,br,1 H,OH),9.44(s,br,1 H,OH),8.30(d,J=7.8 Hz,1 H),7.93(d,J=7.8 Hz,1 H),7.50(d,J=1.6 Hz,1 H),7.46(d,J=7.8 Hz,1 H),7.39~7.33(m,2 H),6.86(d,J=8.2 Hz,1 H)。13C NMR(100 MHz,DMSO-d6):δ168.0,151.1,149.4,146.2,134.5,126.8,125.3,122.7,122.5,119.9,116.6,114.4。ESI-HRMS:C13H9NO2SNa[M+Na]+,理论值266.025 2,实验值266.026 6。
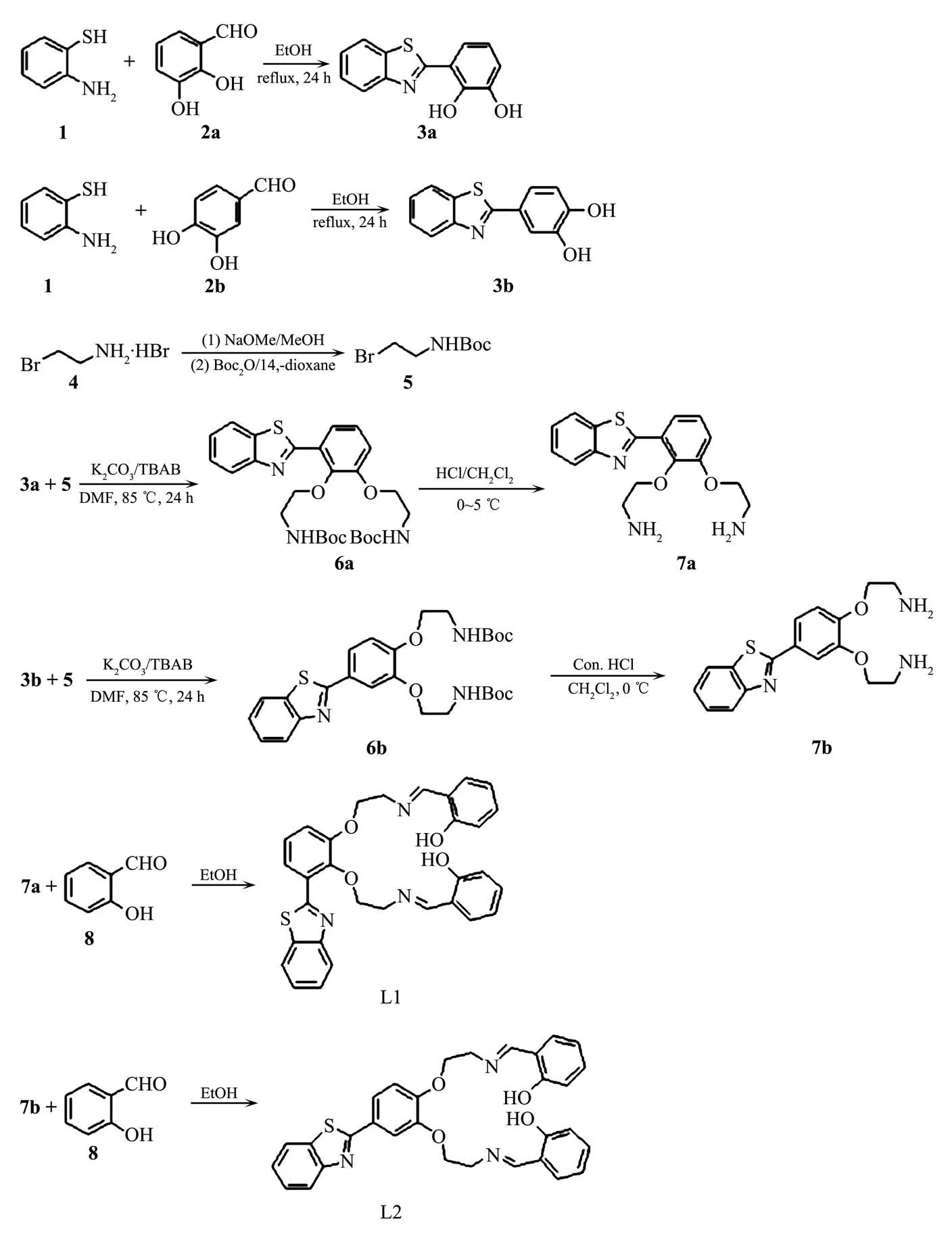
Scheme 1 Synthesis of the chelators
4-(苯并[d]噻唑-2-基)-1,2-苯二酚(3b):3b 的合成方法与3a 相同(3.84 g,79%)。1H NMR(400 MHz,DMSO-d6):δ11.5(s,br,1 H,OH),9.67(s,br,1 H,OH),8.11(d,J=7.8 Hz,1 H),8.03(d,J=8.2 Hz,1 H),7.55~7.48(m,2 H),7.42(t,J=7.8 Hz,1 H),6.95(dd,J=7.8,1.2 Hz,1 H),6.81(t,J=8.2 Hz,1 H)。13C NMR(100 MHz,DMSO-d6):δ166.9,151.8,146.7,146.1,134.2,127.0,125.6,122.5,120.0,118.9,118.7,118.3。ESIHRMS:C13H10NO2S[M+H]+,理论值244.043 2,实测值244.044 4。
Boc保护的2-溴乙胺(5):2-溴乙胺氢溴酸盐(4)(8.2 g,40 mmol)溶于干燥的甲醇(40 mL)中,向溶液中加入甲醇钠(2.16 g,40 mmol),室温搅拌30 min后,加入1,4-二氧六环(120 mL)和二碳酸二叔丁酯(6.96 g,40 mmol)以及三乙胺(4.04 g,40 mmol)。混合物在室温下继续搅拌2 h。滤去溴化钠,旋干,残余物溶于二氯甲烷(600 mL),水洗3次。有机相经无水硫酸钠干燥后,旋干,残余物经硅胶柱色谱(VEtOAc∶Vhexane=1∶3)纯化后得到5(7.62 g,85%)。
Boc保护的 2,2′-(3-(苯并[d]噻唑-2-基)-1,2-苯基)二氧乙基二胺(6a):化合物 3a(2.43 g,10 mmol)与化合物 5(4.28 g,20 mmol)溶于 DMF(40 mL)中,向溶液中加入K2CO3(2.76,20 mmol)与四丁基溴化铵(1.61 g,5 mmol)。混合物在85℃下搅拌24 h。滤去固体物后,滤液用乙酸乙酯(200 mL)稀释。用饱和食盐水洗涤3次,有机相经无水硫酸钠干燥后,旋干,残余物经硅胶柱色谱(VEtOAc∶Vhexane=1∶3)纯化得6a(3.87 g,73%)。1H NMR(400 MHz,DMSO-d6):δ8.07(d,J=5.4 Hz,1 H),8.04(d,J=5.4 Hz,1 H),7.93(d,J=4.8 Hz,1 H),7.51(t,J=5.4 Hz,1 H),7.43(t,J=5.4 Hz,1 H),7.24~7.14(m,2 H),7.01(s,br,1 H,NH),6.92(s,br,1 H,NH),4.15(t,J=4.0 Hz,2 H),4.05(t,J=3.6 Hz,2 H),3.45~3.35(m,4 H),1.36(s,9 H),1.33(s,9 H)。13C NMR(100 MHz,DMSO-d6):δ170.7,162.7,156.1,156.0,152.1,146.4,136.0,126.73,126.70,125.7,124.7,123.1,122.2,120.7,116.5,78.2,71.8,68.0,60.2,40.1,28.7。ESI-HRMS:C27H35N3O6SNa[M+Na]+,理论值552.214 4,实验值 552.216 8。
Boc保护的 2,2′-(4-(苯并[d]噻唑-2-基)-1,2-苯基)二氧乙基二胺(6b):6b的合成方法与6a相同(4.08,77%)。1H NMR(400 MHz,CDCl3):δ8.10(d,J=8.2 Hz,1 H),7.86(d,J=7.8 Hz,1 H),7.73(s,1 H),7.65~7.56(m 1 H),7.46(t,J=5.4 Hz,1 H),7.35(t,J=7.6 Hz,1 H),6.98(d,J=8.2 Hz,1 H),5.28(s,br,2 H,2 NH),4.19(t,J=4.6 Hz,2 H),4.12(t,J=5.2 Hz,2 H),3.51(s,4 H),1.45(s,18 H)。13C NMR(100 MHz,CDCl3):δ167.4,146.2,154.1,134.9,132.8,127.6,126.3,125.0,124.8,122.9,122.1,121.5,119.7,94.0,82.4,79.7,69.6,69.1,40.1,28.4。ESI-HRMS:C27H35N3O6SNa[M+Na]+,理论值552.214 4,实验值552.212 8。
2,2′-(3-(苯并[d]噻唑-2-基)-1,2-苯基)二氧乙基二胺(7a):化合物 6a(2.65 g,5 mmol)溶于二氯甲烷(15 mL),冷却至0℃,在此温度和搅拌下,滴加浓盐酸(4.1 mL),完毕后继续搅拌20 min,过滤,滤饼用蒸馏水溶解后,用1 mol·L−1的NaOH溶液调pH值到9~10。用二氯甲烷萃取该混合物,有机相用饱和食盐水洗涤3次,经无水硫酸钠干燥后旋干,得7a(1.45 g,88%)。1H NMR(400 MHz,DMSO-d6):δ8.11(d,J=4.8 Hz,1 H),8.03(d,J=4.8 Hz,1 H),7.93(d,J=2.8 Hz,1 H),7.51(t,J=4.8 Hz,1 H),7.43(t,J=4.8 Hz,1 H),7.22(s,2 H),4.12(s,2 H),4.04(s,2 H),3.04(s,2 H),2.98(s,2 H)。13C NMR(100 MHz,DMSO-d6):δ162.2,152.4,152.1,146.7,135.9,126.8,125.7,124.8,123.0,122.4,120.4,116.5,75.3,70.9,55.3,42.0,41.1。ESIHRMS:C17H20N3O2S[M+H]+,理论值 330.127 6,实验值330.126 0。
2,2′-(4-(苯并[d]噻唑-2-基)-1,2-苯基)二氧乙基二胺(7b):7b 的合成方法与 7a 相同(1.43 g,87%)。1H NMR(400 MHz,DMSO-d6):δ8.06(d,J=7.8 Hz,1 H),7.99(d,J=7.8 Hz,1 H),7.64(d,J=1.5 Hz,1 H),7.57(dd,J=8.2,1.5 Hz,1 H),7.49(t,J=7.8 Hz,1 H),7.39(t,J=7.8 Hz,1 H),7.10(d,J=8.2 Hz,1 H),4.05~3.95(m,4 H),2.94~2.85(m,4 H)。13C NMR(100 MHz,DMSO-d6):δ167.6,154.0,151.9,149.2,134.7,127.0,126.2,125.6,122.9,122.6,121.6,114.2,112.2,72.0,71.7,41.4,41.0。ESI-HRMS:C17H20N3O2S[M+H]+,理论值330.127 6,实验值330.129 0。
N,N′-双(水杨醛)缩-2,2′-(3-(苯并[d]噻唑-2-基)-1,2-苯基)二氧乙基二胺(L1):化合物 7a(0.66 g,2 mmol)溶于无水乙醇(10 mL)中,向溶液中加入水杨醛(0.488 g,4 mmol)。反应混合物在室温下搅拌2 h,得大量黄色结晶,过滤,滤饼用冷乙醇洗涤3次,真空干燥得产物 L1(0.91 g,85%)。1H NMR(400 MHz,DMSO-d6):δ13.47(s,br,1 H,OH),13.36(s,br,1 H,OH),8.67(s,1 H,CH=N),8.47(s,1 H,CH=N),7.97(d,J=5.2 Hz,1 H),7.94(d,J=4.8 Hz,1 H),7.72(d,J=5.2 Hz,1 H),7.48~7.43(m,2 H),7.38~7.27(m,5 H),7.23~7.19(m,1 H),6.94~6.87(m,4),4.39(t,J=3.0 Hz,2 H),4.33(t,J=3.0 Hz,2 H),4.03(t,J=3.0 Hz,2 H),3.87(t,J=3.0 Hz,2 H)。13C NMR(100 MHz,DMSO-d6):δ167.74,167.70,162.4,161.1,161.0,152.2,146.1,136.8,135.9,132.93,132.85,132.2,129.6,126.7,125.6,123.0,121.9,120.6,119.9,119.1,117.7,117.0,116.9,116.5,72.1,68.6,59.8,58.2。ESI-HRMS:C31H27N3O4SNa[M+Na]+,理论值560.162 0,实验值560.164 4。
N,N′-双(水杨醛)缩-2,2′-(3-(苯并[d]噻唑-2-基)-1,2-苯基)二氧乙基二胺(L2):L2的合成方法与L1相同 (0.85 g,79%)。1H NMR(400 MHz,DMSO-d6):δ13.35(s,br,1 H,OH),13.32(s,br,1 H,OH),8.52(s,2 H,2 CH=N),8.03(d,J=7.8 Hz,1 H),7.98(d,J=7.8 Hz,1 H),7.64(s,1 H),7.60~7.55(m,1 H),7.48(d,J=7.8 Hz,1 H),7.42~7.34(m,3 H),7.29(t,J=7.8 Hz,2 H),7.16~7.09(m,1 H),6.89~6.81(m,4 H),4.35~4.24(m,4 H),3.87(s,4 H)。13C NMR(100 MHz,DMSO-d6):δ167.8,161.0,154.0,151.6,148.9,134.8,132.1,126.9,126.6,125.6,123.0,122.6,121.9,119.1,119.0,118.96,117.0,114.9,113.2,69.0,68.6,58.0,57.9。ESI-HRMS:C31H27N3O4SNa[M+Na]+,理论值 560.162 0,实验值560.159 8。
1.3 生物活性测试
1.3.1 Aβ比浊度测定
在 198 µL 的 HEPES 缓 冲 溶 液(50 mmol·L−1HEPES,150 mmol·L−1NaCl,pH=7.4)中 加 入 20µmol·L−1的Aβ1~40和40 µmol·L−1的锌离子或铜离子,在室温下反应5 min后,加入2µL的L1、L2以及L3的DMSO储备液,使螯合剂在样品溶液中的浓度为40 µmol·L−1,DMSO含量1%。在37 ℃条件下孵育24 h,每个样品溶液分别移至96孔板的一个孔中,样品溶液的浊度通过405 nm处的吸光度获得。相同条件下,以Aβ1~40溶液作对照,相同实验做3组,以计算误差。
1.3.2 Bicinchoninic acid(BCA)蛋白测定
样品的配制方法与1.3节中的相同,但Aβ1~40的浓度为 100 µmol·L−1,金属离子的浓度与 Aβ1~40相同,每个含有Zn2+(或Cu2+)的样品准备12份,样品溶液在37℃下孵育48 h后,只含Aβ1~40的样品和从含有Zn2+或Cu2+的样品中取出3份,在14 000 r·min−1转速下高速离心20 min。取出上层清液用Micro BCA protein Assay Kit测定蛋白浓度。另外剩下的9份Zn2+(或Cu2+)的样品分成3组,分别加入与金属等物质的量的螯合剂L1和L2以及L3,然后再在37℃并不断摇动下孵育24 h,样品在 14 000 r·min−1转速下高速离心20 min。取出上层清液用Micro BCA protein Assay Kit测定蛋白浓度。相同实验做3组,以计算误差。
1.3.3 H2O2的测定
H2O2的测定由HRP/Amplex Red实验完成。将试剂按以下次序加入到96孔板中,使其体积为100µL,样品溶液中含有 Aβ1~40(0.2 µmol·L−1)和铜离子(0.2或0.4 µmol·L−1)以及螯合剂L1、L2、L3(0.2或0.4µmol·L−1)、抗坏血酸(10 µmol·L−1),用只含有 Aβ1~40和铜离子的溶液作为对照组。样品在室温下孵育30 min后,向每个孔中加入50µL新配制的工作液,其中含有0.1µL的Amplex Red(Sigma-Aldrich)和0.2 U·mL−1的 HRP(Sigma-Aldrich),继续在室温下孵育30 min,荧光光谱用一台瑞典生产的TECAN多功能酶标仪(λex=530 nm,λem=590 nm)测得。相同实验做3组,以计算误差。
1.3.4 MTT实验分析细胞毒性
PC12细胞在含10%胎牛血清的DMEM培养基中,置于37℃、5%(%)CO2培养箱中培养。细胞毒性检测时,将对数生长期的PC12细胞接种于12孔板中,孵育过夜后,将实验分为正常细胞对照组、Aβ1~40-M2+(Zn2+,Cu2+)组、Aβ1~40-M2+(Zn2+,Cu2+)+不同待测螯合剂(L1,L2,L3)组,按照分组,分别加入不同浓度的待测样品(20 µmol·L−1)、10 µmol·L−1Aβ1~40和 20µmol·L−1的铜离子继续孵育48 h后,加入终浓度为0.5 mg·mL−1的MTT在37 ℃孵育2 h。最后去掉上清液,用DMSO室温裂解细胞,溶解甲臜盐,然后在酶标仪570 nm处检测吸光度值。
2 结果与讨论
2.1 抑制Aβ聚集
已有的研究表明,金属离子如Cu2+和Zn2+等都能促进Aβ聚集,而金属螯合剂则能阻止或减弱这种聚集作用[30-31]。本实验通过Aβ比浊度分析来检测螯合剂L1和 L2在金属离子(Zn2+、Cu2+)诱导的 Aβ1~40聚集的干预作用,结果见图3。
图3结果显示,在没有金属离子和螯合剂存在下,Aβ1~40聚集很低,在405 nm的吸光度只有0.006,而有螯合剂(L1、L2、L3)存在,没有金属离子时,与纯的Aβ1~40相比,吸光度稍有变化,但都小于0.009。当有Zn2+或Cu2+存在时,样品溶液的比浊度大幅增加,吸光度分别达到0.062和0.039,这与文献报道的在pH=7.4条件下Zn2+比Cu2+更能促进Aβ聚集的结果相符[24]。螯合剂L1和L2虽然不能完全抑制Zn2+或Cu2+诱导的Aβ1~40聚集,但抑制效果非常显著。L1和L2分别使含锌的样品吸光度下降到0.021和0.025;分别使含铜的样品吸光度下降到0.017和0.020。不含苯并噻唑而螯合部位相同的螯合剂L3虽然也能抑制Aβ1~40聚集,但其效果比L1和L2差。
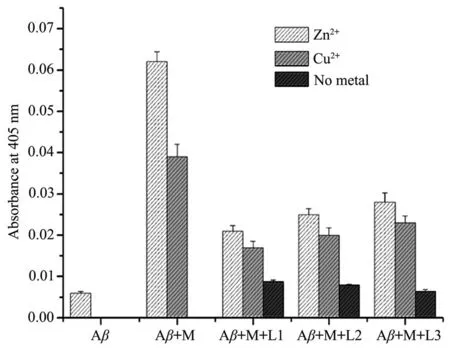
图3 抑制Aβ聚集实验中,螯合剂、Zn2+、Cu2+、Aβ1~40(20 µmol·L−1)在37 ℃、pH 7.4的缓冲溶液中孵育24 h后样品溶液的比浊度(A405)Fig.3 Turbidity(absorbance at 405 nm)of Aβ1~40(20 µmol·L−1)solutions without or with Zn2+,Cu2+ion in the absence and presence of chelators after incubation at 37℃and pH 7.4 for 24 h
考察螯合剂对Aβ1~40聚集的抑制作用,也可用micro bicinchoninic acid(BCA)蛋白标定试剂盒测试法测定样品溶液上层清液中可溶性的Aβ百分含量来实现。测量可溶性的Aβ百分含量时样品的配制方法与比浊度分析相同,但可溶的Aβ以及相应的金属离子和螯合剂浓度都是比浊度分析时的5倍。结果见图4。
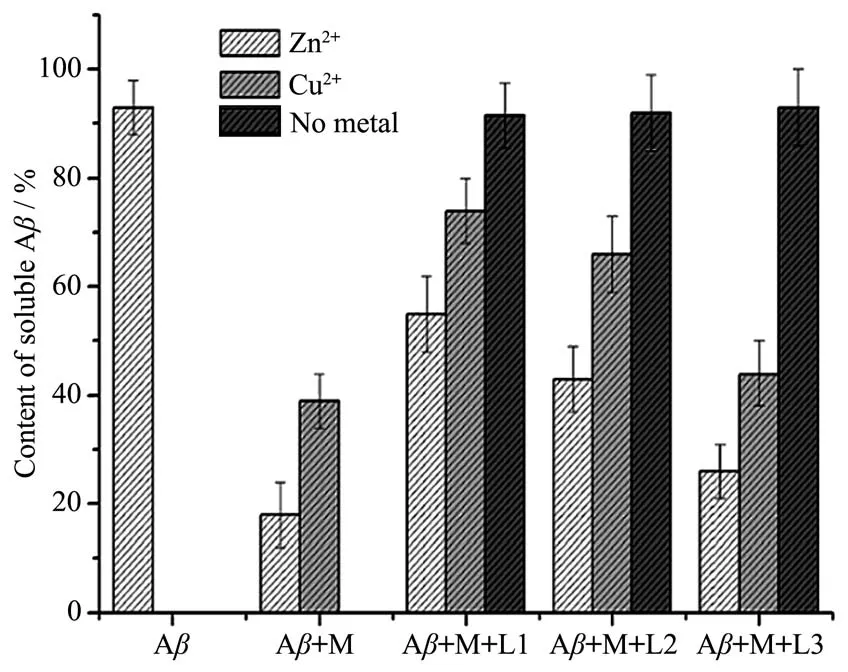
图4 Zn2+、Cu2+与Aβ1~40(100 µmol·L−1)在37 ℃、pH 7.4下孵育24 h后,再加入螯合剂在相同条件下又共同孵育24 h后,上层清液中Aβ的百分含量Fig.4 Percentage of soluble Aβ1~40after Zn2+,Cu2+and Aβ1~40(100 µmol·L−1)incubated at 37 ℃,pH 7.4 for 24 h,then the mixture with chelator incubated under same conditions for 24 h
在没有金属离子存在下,无论样品中是否含有螯合剂,游离的Aβ百分含量是93%左右,但是在Zn2+或Cu2+存在,没有螯合剂存在时,其可溶性Aβ百分含量分别下降到18%和46%。当Aβ与金属离子的样品在37℃下孵育48 h后,再加入螯合剂又共同孵育24 h后,含有所合成的螯合剂L1和L2的样品上层清液中可溶性Aβ的百分含量显著增加,L1和L2分别使含锌离子的上层清液中可溶性Aβ的百分含量上升到55%和43%;分别使含铜离子的上层清液中可溶性Aβ的百分含量上升到74%和66%。而当螯合剂是L3时,可溶性Aβ的百分含量也有一定程度增加,但增幅不大,使含锌或铜离子的上层清液中可溶性Aβ的百分含量分别上升到26%和44%。这可能是含有苯并噻唑的螯合剂能识别Aβ,与Aβ-M加合物相互作用,从而将金属离子从Aβ-M加合物中螯合出来,促使聚合的Aβ解聚[28],而L3因不能识别Aβ,不能很好地与Aβ-M加合物相互作用,因此其促使聚合的Aβ解聚的作用并不明显。
2.2 控制Aβ-Cu的H2O2产物形成
Aβ肽与具有氧化−还原活性的金属离子(如Cu2+)相互作用会形成活性氧(reactive oxygen species,ROS)(如H2O2)和与Aβ神经毒性相联系的氧化应激[32-33],因此,从理论上讲,具有识别Aβ功能的金属螯合剂可以控制活性氧的形成。L1和L2对Aβ-Cu2+形成H2O2的影响用HRP/Amplex Red实验测定。其结果如图5所示。
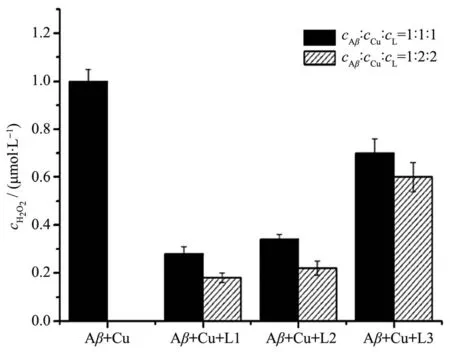
图5 Amplex Red分析中在Aβ、Cu2+、螯合剂、抗坏血酸钠存在下产生的标准过氧化氢的量Fig.5 Normalized amounts of H2O2produced in presence of Aβ,Cu,the chelating agent,and sodium ascorbate,as determined by the Amplex-Red assay
在还原条件下,Cu2+-Aβ与氧气作用产生H2O2,加入我们合成的螯合剂L1和L2后,能清除绝大部分(80%以上)由Cu2+-Aβ产生的H2O2,这说明螯合剂L1和L2能有效控制Aβ-Cu产生H2O2的量。而不能识别Aβ的螯合部位相同的螯合剂L3虽然也能使H2O2产生量减少,但其作用没有L1和L2明显,只能清除40%左右的H2O2。这可能是因为螯合剂L1(或L2)被苯并噻唑功能化后,增强了与Aβ相互作用的能力,从而更易接触Cu2+-Aβ加合物,将Cu2+从Cu2+-Aβ中螯合出来,从而阻止了H2O2产生。这与文献报道的结果相符[34]。
2.3 抑制Aβ诱导的神经毒性
上文的结果表明,螯合剂L1和L2能有效抑制金属Zn2+、Cu2+诱导的Aβ聚集,同时具有抗氧化能力,因此,我们进一步利用PC12细胞株——一个常用的神经作用细胞株(来源于一种可移植的鼠嗜铬细胞瘤),来检测在细胞水平上,螯合剂L1或L2对Aβ1~40聚集造成的神经细胞活力损伤的保护作用下由单体到聚合体进程变化,结果见图6。
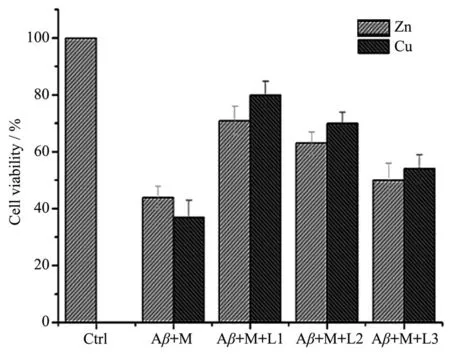
图6 MTT法测定Aβ1~40在金属离子与螯合剂存在与否时对PC12细胞的神经毒性Fig.6 Neurocytotoxicity of Aβ1~40in the absence or presence of metal ions and chelators toward PC12 cells determined by MTT assay
图 6 的结果表明,当 10 µmol·L−1的 Aβ1~40与 20µmol·L−1的Zn2+作用PC12细胞48 h后,PC12细胞存活率下降到 44%;而 10 µmol·L−1的 Aβ1~40与 20µmol·L−1的Cu2+作用PC12细胞48 h后,PC12细胞存活率下降到37%。当10 µmol·L−1的Aβ1~40、20 µmol·L−1的 Zn2+与 20 µmol·L−1的螯合剂 L1 或 L2 作用PC12细胞48 h后,细胞存活率分别为71%和63%;20 µmol·L−1的螯合剂 L1 或 L2 与 10 µmol·L−1的Aβ1~40、20 µmol·L−1的Cu2+作用PC12细胞48 h后,细胞存活率分别提升到80%与70%。螯合剂L3也具有抑制Aβ诱导的神经毒性的作用,但效果比螯合剂L1 和 L2 差,当 10 µmol·L−1的 Aβ1~40、20 µmol·L−1的Zn2+或20 µmol·L−1的Cu2+与20 µmol·L−1的螯合剂L3作用PC12细胞48 h后,细胞存活率分别提升到50%和54%。实验结果表明,相比较单一功能螯合剂,苯并噻唑功能化的螯合剂L1和L2更能抑制Aβ1~40聚集介导的神经毒性并提高细胞存活率。
3 结 论
合成了2种未见文献报道的苯并噻唑功能化的金属螯合剂L1和L2。通过比浊度分析、BCA蛋白测定考察了这2种螯合剂抑制金属Zn2+与Cu2+诱导的 Aβ1~40聚集的性能;通过HRP/Amplex Red实验测定了它们控制Cu-Aβ加合物产生H2O2的量;通过MTT实验检测其存在与否对金属离子诱导的Aβ1~40聚集对PC12细胞神经细胞毒性的影响,并与非苯并噻唑功能化的螯合部位相同的螯合剂进行了比较。发现这2种螯合剂都能有效抑制Zn2+与Cu2+诱导的Aβ1~40聚集;大幅减少Cu-Aβ加合物产生H2O2的量;有效抑制Zn2+、Cu2+诱导Aβ聚集而产生的神经细胞毒性并大幅提高细胞存活率。作为对比,非苯并噻唑功能化但螯合部位相同的螯合剂虽然在相同条件下也有作用,但其活性远低于苯并噻唑功能化的金属螯合剂。这一研究表明,对照单一功能的金属螯合剂,苯并噻唑功能化的金属螯合剂更有可能成为治疗AD的药物。
Supporting information is available at http://www.wjhxxb.cn
猜你喜欢
环境科学研究(2022年10期)2022-10-19
生殖医学杂志(2022年10期)2022-10-19
中南民族大学学报(自然科学版)(2022年3期)2022-05-08
小作家报·教研博览(2022年11期)2022-04-02
中国土壤与肥料(2021年5期)2021-12-02
中国农业气象(2021年12期)2021-11-30
中国瓜菜(2020年9期)2020-11-30
中小学德育(2020年11期)2020-03-18
农业与技术(2018年3期)2018-03-21
教育界·上旬(2016年12期)2017-05-25