紫苏醇芳基钌配合物的合成及抗肿瘤性能
2020-07-20 02:06薛旭玲刘红科
无机化学学报 2020年7期
吴 健 陶 钦 葛 超 薛旭玲 钱 勇 刘红科
(南京师范大学化学与材料科学学院,南京 210023)
0 引 言
以顺铂为代表的金属配合物具有良好的药理学特性,引起了人们对金属抗癌药物的极大兴趣[1-3],但铂类药物毒副作用大、固有或获得性耐药性等不足大大限制了铂类药物的应用,因此新型钌、铱等金属抗癌药物逐渐走入人们的视野[4-5]。其中,半三明治有机金属配合物因其化学结构的多样性和易于控制的芳烃或环戊二烯基的疏水性而在化学治疗研究中极为突出[6-8]。此外,芳烃或环戊二烯基不仅有助于提高细胞对金属药物的摄取,而且对生物靶标的功能模式和配合物的动力学惰性也具有重要作用[9-11]。Sadler等报道此类金属配合物不同于铂类药物的多种作用机理,具有毒性小、耐药性低及易代谢等独特的优势[12-13]。在此类有机金属配合物中,含有各种螯合配体的Ru (Ⅱ)-芳烃配合物表现出优异的化学可修饰性,例如将芳基钌配合物与其他生物活性分子结合,可以轻松地修改其化学结构,以调节其物理化学性质和生物活性[14-18]。
天然产物因其可修饰的化学结构、丰富的生物活性以及潜在多功能靶点等优势,也得到了人们的广泛关注,目前有科研工作者也将其引入金属抗癌药物以提高药物的生物活性[19-21]。其中,紫苏醇作为一种治疗及预防癌症的单萜类口服药物,曾经进入临床二期试验[22-23],在乳腺癌(IC50≈500 µmol·L−1)[24]、肺癌(IC50≈1 000 µmol·L−1)[25]等多种肿瘤细胞具有一定的防癌及抗癌作用。紫苏醇通过在血清中被氧化为紫苏酸和脱水紫苏酸来发挥其抗癌作用[26]。此外,紫苏醇还可以抑制血管生成[27],诱导MCF-7细胞阻滞在G0/G1期,引起p21Cip1/Wafl水平的上调。但因紫苏醇口服给药生物利用度低、给药剂量大,高剂量的摄入会引起肠道毒性,如导致患者恶心、疲劳和呕吐等副作用。因此,开发一种抗肿瘤活性强、毒副作用小的紫苏醇药物具有广泛的应用前景。
在本研究中,我们对紫苏醇进行化学修饰,合成了基于紫苏醇的新型芳基钌配合物,并通过1H NMR、ESI-MS及元素分析进行了结构表征。采用MTT法评估了配合物对多种肿瘤细胞株的抗增殖能力,并利用UV-Vis分光光度计及荧光光谱仪检测了配合物与重要生物分子如DNA、BSA及HSA的相互作用,同时利用流式细胞术通过细胞内活性氧生成、细胞周期及细胞凋亡实验探讨了配合物诱导细胞死亡的作用机理。
1 实验部分
1.1 试剂及仪器
实验所用的化学药品和溶剂均为商用分析纯,无需进一步纯化即可直接使用。4,4′-dimethyl-2,2′-bipyridine购自晚晴化学品公司,根据文献方法制备了化合物 4′-methyl-2,2′-bipyridine-4-carboxylic acid[28]和二聚体[Ru(η6-p-cym)Cl2]2[29]。4-二甲氨基吡啶(DMAP)、1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDCI)等药品购自阿拉丁试剂有限公司。
细胞培养基Dulbecco′s Modified Eagle Medium(DMEM,Invitrogen)、双抗(含有青霉素和链霉素的混合液)和胎牛血清等购自Gibco公司。PBS缓冲溶液、胰蛋白消化酶-EDTA等生物试剂购自南京凯基生物科技有限公司。紫苏醇购自北京华威锐科化工有限公司。所有的反应均在氩气氛围下进行。
1H NMR使用Bruker AvanceⅡ400 MHz光谱仪检测。电喷雾电离质谱(ESI-MS)通过使用Mass Lynx系统的Thermo LCQ-FLEET质谱仪测定。在PerkinElmer 240C元素分析仪上测量C,H和N的含量。UV-Vis数据采用PerkinElmer Lambda 365紫外−可见光谱仪进行收集。荧光光谱仪采用Edinburgh FS5型号。使用的多功能酶标仪型号为LabServ K3,流式细胞仪型号为BD FACSverse。
1.2 紫苏醇化学修饰配体及配合物的合成
1.2.1 紫苏醇化学修饰配体(L)的合成
将 4′-methyl-2,2′-bipyridine-4-carboxylic acid(82.20 mg,0.54 mmol)、紫苏醇(173.5 mg,0.81 mmol)和DMAP(131.9 mg,1.08 mmol)的DMF溶液在氩气和冰浴条件下搅拌15 min,然后加入EDCI(207.0 mg,2.0 mmol),室温搅拌24 h。TLC监测至反应结束后旋干溶剂,将粗产品用乙酸乙酯(3×20 mL)萃取,用水、盐水洗涤,并通过快速柱色谱法(石油醚−乙酸乙酯)纯化,产物为无色油状物(100.3 mg,53.3%)。1H NMR(400 MHz,DMSO-d6):δ8.89(dd,J=5.0,0.8 Hz,1H),8.82(dd,J=1.6,0.8 Hz,1H),8.59(d,J=4.9 Hz,1H),8.27(s,1H),7.89(dd,J=5.0,1.7 Hz,1H),7.34(ddd,J=4.9,1.6,0.7 Hz,1H),5.88(s,1H),4.78(s,2H),4.72(s,2H),2.44(s,3H),2.21~2.06(m,4H),1.97(dd,J=13.8,2.4 Hz,1H),1.86~1.77(m,1H),1.71(s,3H),1.45(tt,J=12.7,8.5 Hz,1H)。13C NMR(101 MHz,DMSO-d6):δ164.91,156.99,154.46,150.99,149.74,149.44,148.76,138.55,132.56,126.13,125.99,123.21,121.81,119.60,109.52,69.52,41.22,30.27,27.24,26.28,21.18,21.06。ESI-MS(m/z):[L+H]+理论值 349.44,实验值349.32。
1.2.2 配合物[Ru(η6-p-cym)(bpy-POH)Cl]Cl(Ru-L)的合成
将二聚体[Ru(η6-p-cym)Cl2]2(25.0 mg,0.04 mmol)和配体L(28.5 mg,0.08 mmol)的二氯甲烷溶液在氩气下回流12 h,TCL监测至反应结束后旋干溶剂,并将粗产物通过快速柱色谱法(二氯甲烷−甲醇)纯化,产物为浅红色粉末(39.4 mg,75.2%)。1H NMR(400 MHz,DMSO-d6):δ9.74(d,J=5.9 Hz,1H),9.40(d,J=5.8 Hz,1H),8.92(s,1H),8.81(s,1H),8.09(dd,J=5.9,1.6 Hz,1H),7.69(d,J=5.7 Hz,1H),6.25(dd,J=9.1,6.3 Hz,2H),6.02(dd,J=9.4,6.3 Hz,2H),5.91(s,1H),4.85(s,2H),4.73(s,2H),2.60(s,4H),2.18(s,7H),2.02~1.92(m,1H),1.87~1.78(m,1H),1.73(s,3H),1.52~1.42(m,1H),0.95(d,J=6.9 Hz,6H)。13C NMR(101 MHz,CDCl3):δ163.09,158.50,156.13,155.14,153.38,151.81,149.34,140.01,131.51,129.58,128.97,127.93,126.98,126.28,123.94,121.65,108.98,104.93,104.44,70.89,40.58,31.18,30.47,27.18,26.53,22.29,22.22,21.56,20.80。ESI-MS(m/z):[Ru-L−Cl]+理论值619.18,实验值619.33。元素分析:按C32H38Cl2N2O2Ru计算值(%):C 58.71,H 5.85,N 4.28。实验值(%):C 59.04,H 5.91,N 4.16。
1.3 细胞毒性研究
原料紫苏醇(perillol)、紫苏醇化学修饰配体L、配合物Ru-L及其对照药物顺铂的细胞毒性实验采用MTT(3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide)法测定,选择人源肿瘤细胞系卵巢癌(A2780)、卵巢癌的顺铂耐药株(A2780/DDP)、乳腺癌(MCF-7)和正常人源细胞肺成纤维细胞(HLF)进行测试。细胞在DMEM(Dulbecco改良的Eagle培养基,Gibco BRL)中培养,其中含有10%FBS(胎牛血清,Gibco BRL),100 µg·mL−1链霉素和100 U·mL−1青霉素(Gibco BRL)。将处于对数生长期的4种细胞系以每孔5 000个细胞的初始密度接种到96孔细胞培养板中,在5%(V/V)CO2、310 K条件下培养24 h后,加入待测药物(药物溶于1%(V/V)DMSO的DMEM培养基溶液),继续孵育48 h。在每孔加入20µL(5 mg·mL−1)的MTT后继续孵育4 h,然后吸去培养基,加入150µL DMSO,振荡10 min后,使用LabServ K3酶标仪读取492 nm处吸光度值。测得的IC50值是平均值±SEM。
1.4 配合物的水解
通过紫外可见光谱法测定配合物Ru-L的水解常数及半衰期。制备的纯化样品在H2O(含10%(V/V)DMSO)中的浓度为50 µmol·L−1。在298 K下,24 h内以选定的波长(250~550 nm)在15或30 min的间隔内记录吸光度。将吸光度随时间变化的图拟合为适当的等式。(A=C0+C1e−kt,其中C0和C1是计算机拟合的常数,A是配合物与时间相对应的吸光度),用于拟合一级反应动力学,以计算水解速率常数K和半衰期t1/2。
1.5 配合物与CT-DNA的相互作用
使用配合物Ru-L的DMSO储备液在PBS(10 mmol·L−1,含有 10 mmol·L−1NaCl,pH 7.4)中稀释并进行DNA结合实验。用PBS将CT-DNA稀释至所需的浓度,并通过UV-Vis光谱测定CT-DNA在260 nm处吸光度值,确定CT-DNA的浓度(ε=6 600 L·mol−1·cm−1)。同时测定其在280 nm处的吸光度值,A260/A280的比值约为1.87,表明DNA溶液不含蛋白质,对DNA滴定实验没有干扰。固定每个样品中配合物的浓度,加入CT-DNA的浓缩储备液,其中在参比池中每次都要添加等浓度CT-DNA,以最大程度地减少由于DNA在260 nm吸收引起的实验干扰。将样品在生理条件下(PBS,10 mmol·L−1,含有 10 mmol·L−1NaCl,pH 7.4)室温孵育 10 min以达到结合平衡,然后记录配合物Ru-L在302 nm处的吸光度。将配合物Ru-L与CT-DNA的相互作用拟合到Benesi-Hildebrand[30]方程,以计算结合常数Kb。
1.6 配合物与BSA/HSA的相互作用
使用固定浓度的BSA或HSA进行荧光猝灭滴定实验研究配合物与BSA或HSA的相互作用。在PBS(10 mmol·L−1,含有 10 mmol·L−1NaCl,pH 7.4)中制备BSA或HSA储备溶液并在4℃下保存。每次连续加入配合物Ru-L并在室温下孵育10 min以完成相互作用后,记录不存在和存在配合物Ru-L的情况下 BSA(3.0 µmol·L−1)或 HSA(1.0 µmol·L−1)的荧光发射光谱,配合物Ru-L的浓度区间分别设为0~20µmol·L−1和0~5 µmol·L−1。其中BSA和HSA激发波长分别为280和278 nm,测定BSA或HSA溶液在334或307 nm处的荧光强度。
1.7 细胞凋亡检测
将处于对数生长期的A2780细胞以每孔2×104个细胞的初始密度接种到6孔细胞培养板中。在5%(V/V)CO2、310 K条件下培养24 h后,加入不同浓度(0、11、22和 33 µmol·L−1)的待测药物(含有1%(V/V)DMSO的DMEM培养基溶液)。继续孵育24 h后,消化并收集细胞。用PBS洗涤2次,加入500µL的binding buffer重悬细胞,并加入5µL的Annexin VFITC,5 min后加入5µL的propidium iodide混匀,室温避光静置15 min,随后立即使用BD FACSverse流式细胞仪检测,使用FlowJo 7.6软件处理数据。
1.8 细胞周期阻滞检测
将处于对数生长期的A2780细胞以每孔2×104个的初始密度接种到6孔细胞培养板中。在5%(V/V)CO2、310 K条件下培养24 h后,加入不同浓度(0、11、22 和 33 µmol·L−1)的待 测药物(含 有 1%(V/V)DMSO的DMEM培养基溶液)。继续孵育24h后,消化并收集细胞。用PBS洗涤2次,加入300µL的PBS重悬细胞,向其中缓慢滴加700µL冰乙醇,277 K过夜固定细胞。固定结束后,2 000 r·min−1离心5 min,PBS洗涤,加入提前配好的碘化丙啶染色液(PI/RNase A,9∶1,V/V),室温下避光静置30 min,随后立即使用BD FACSverse流式细胞仪检测,使用FlowJo 7.6软件处理数据。
1.9 细胞内活性氧(ROS)检测
将处于对数生长期的A2780细胞以每孔2×104个的初始密度接种到6孔细胞培养板中,在5%(V/V)CO2、310 K条件下培养24 h后,加入不同浓度(0、11、22和33 µmol·L−1)的待测配合物 Ru-L(含有 1%(V/V)DMSO的DMEM培养基溶液)。继续孵育4h后,收集细胞,用DMEM培养基洗涤2次,并向其中加入10 µmol·L−1活性氧探针H2DCFDA,避光继续孵育20 min。PBS洗涤2次后收集细胞并用PBS重悬细胞。随后立即使用BD FACSverse流式细胞仪检测,激发波长为488 nm,发射波长为510~540 nm,使用FlowJo 7.6软件处理数据。
2 结果与讨论
2.1 合成和表征
选择具有抗肿瘤活性的天然产物紫苏醇对其进行化学修饰,引入同样具有抗肿瘤活性的芳基钌配合物,希望结合紫苏醇和芳基钌的双重作用,提高配合物Ru-L的抗肿瘤效果。紫苏醇化学修饰配体L是由紫苏醇和4′-methyl-2,2′-bipyridine-4-carboxylic acid在DMF中通过酰胺化反应,并通过柱层析分离提纯制得。芳基二聚体[Ru(η6-p-cym)Cl2]2是由RuCl3·3H2O与α-松油烯在甲醇中制备得到。配合物Ru-L由芳基二聚体[Ru(η6-p-cym)Cl2]2与配体L直接在二氯甲烷中反应,并通过柱层析分离提纯制得(如图1所示)。利用1H NMR、13C NMR、ESI-MS及元素分析对配体L和配合物Ru-L进行了表征。
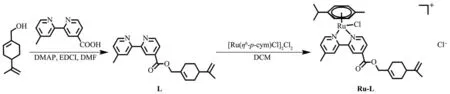
图1 配体L及配合物Ru-L的合成路线Fig.1 Synthetic route of ligand L and complex Ru-L
2.2 细胞毒性研究
通过MTT法研究了紫苏醇、紫苏醇化学修饰配体L以及配合物Ru-L对人源肿瘤细胞株A2780、A2780顺铂耐药株(A2780DDP)、MCF-7和正常细胞HLF 48 h的体外细胞毒性,并以顺铂作为阳性对照药物。从表1可以看出,紫苏醇和配体L对人源肿瘤细胞及正常细胞毒性较小(IC50>200 µmol·L−1),与文献报道数值相一致[21,31],远低于顺铂的细胞毒性。与天然产物、相应配体及[Ru(η6-p-cym)Cl2]2(IC50>200µmol·L−1)相 比[32],配 合 物 Ru-L 对 A2780 细 胞 、A2780/DDP细胞及MCF-7细胞的毒活性显著提高,其IC50值分别为22、40和34 µmol·L−1。同时对人正常细胞HLF毒性较小(IC50>200 µmol·L−1),表明引入天然产物紫苏醇后配合物Ru-L不仅可以显著提高对肿瘤细胞的毒性,而且对正常细胞具有较低的毒副作用。为了探究配合物Ru-L对多种肿瘤细胞株毒活性提高的原因,我们深入研究了配合物Ru-L的水解速率,与重要生物分子如DNA、BSA和HSA的相互作用,以及诱导A2780细胞死亡的作用机理。
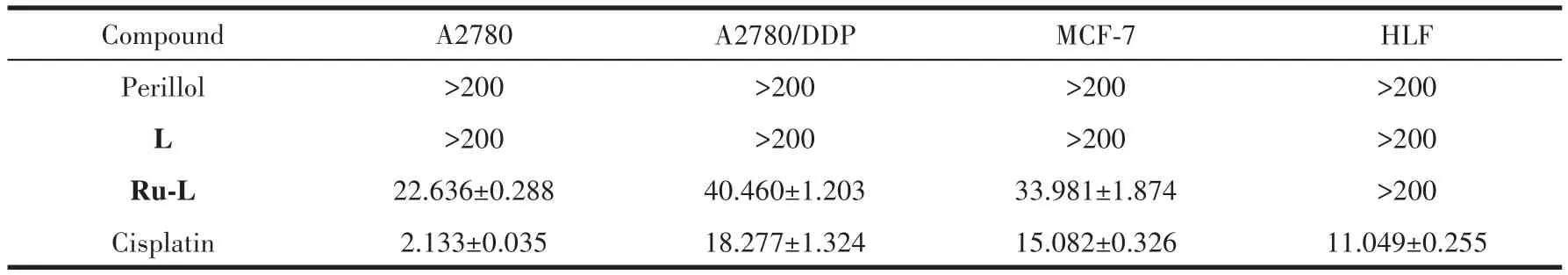
表1 紫苏醇、配体L及配合物Ru-L对不同肿瘤细胞株的48 h的IC50值Table 1 IC50values towards different cells by perillol,L and complex Ru-L for 48 h µmol·L−1
2.3 配合物的水解
芳基金属配合物通过水解来调控其药理学性质,如细胞摄取、细胞毒性及与DNA的相互作用等[33-34],其中配合物水解后可与DNA的碱基发生配位,进而影响DNA的转录及翻译等生理功能[35]。因此我们利用UV-Vis光谱研究配合物Ru-L(50µmol·L−1)在含有10%(V/V)DMSO的水溶液中的水解速率。如图2所示,配合物Ru-L的特征吸收峰为278、296、314、318及398 nm,其中,在278和296 nm处较强的特征峰归属于MLCT/LLCT跃迁,在314和318 nm处的特征峰为离去基团Cl的π轨道跃迁到π*所引起的LLCT跃迁,其中398 nm处的特征峰为Ru原子4d轨道上的电子向配体的π*空轨道MLCT跃迁所致[36-37]。在298 K,100 min之内,配合物Ru-L水溶液中吸收峰发生了明显的变化,在296 nm处的吸收峰减弱(减弱率为7.72%),在278、314、318及398 nm处的吸收峰增强(增强率分别为12.16%、3.63%、8.26%及15.91%),表明配合物发生了水解。利用一级反应动力学方程求导得到ln(C/C0)与时间的关系,其中C0是初始时配合物的浓度,C是对应时间时配合物的浓度。并通过在0~100 min之内的斜率因子计算出配合物的水解速率常数和半衰期分别为1.31 h−1和0.53 h,表明配合物Ru-L在298 K的水溶液环境中具有较快的水解速率[8]。
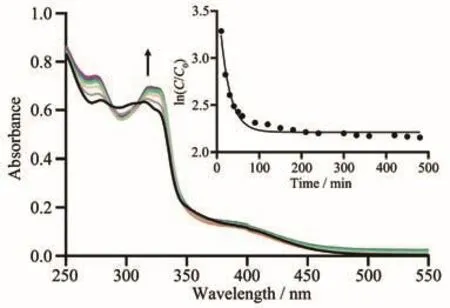
图2 配合物Ru-L在水溶液中的紫外吸收光谱图Fig.2 Ultraviolet absorption spectrum of complex Ru-L in H2O solution
2.4 配合物Ru-L与CT-DNA的相互作用
DNA是芳基金属配合物在肿瘤细胞内潜在靶点之一,研究DNA和芳基金属配合物的相互作用在研究抗肿瘤机制中发挥至关重要的作用。常用紫外−可见光谱研究配合物与小牛胸腺DNA(CT-DNA)可能的结合模式及其结合常数Kb[38]。配合物通过嵌入作用与DNA的碱基对发生电子堆积,配体的π*轨道与DNA碱基对的π轨道发生耦合导致能级下降,耦合后的π*轨道部分被电子填充,使其π-π*跃迁几率减小,从而产生减色效应或红移现象[39]。通过 UV-Vis光谱研究配合物Ru-L(30 µmol·L−1)在含有 10%(V/V)DMSO 的 PBS(10 mmol·L−1,pH 7.4)中的结合常数Kb。如图3所示,在配合物溶液中滴加CTDNA时,在314~318 nm处观察到了明显的减色效应,其中在CT-DNA的浓度为40 µmol·L−1时,配合物的减色效应高达21.36%。
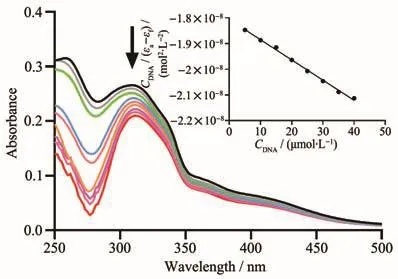
图3 配合物Ru-L在PBS中滴加CT-DNA后的紫外吸收光谱图Fig.3 Ultraviolet absorption spectra of complex Ru-L after the addition of CT-DNA in PBS
将配合物Ru-L与CT-DNA的相互作用拟合到Benesi-Hildebrand方程,以计算结合常数Kb:
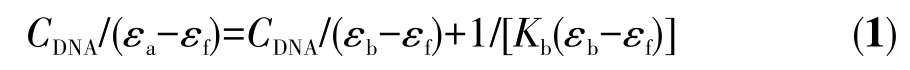
其中CDNA是CT-DNA的浓度,表观吸收系数εa对应于Aobs/CRu-L,εb和εf分别表示结合态和游离态的配合物的消光系数。将CDNA/(εa−εf)与CDNA的关系进行线性拟合,并通过斜率因子与截距的比值计算出配合物与DNA的结合常数Kb为4.37×105L·mol−1,相比之前文献报道[40],配合物Ru-L与DNA具有更强的结合能力,并以嵌入的方式与CT-DNA相互作用,这种结合方式在抗肿瘤药物的设计和开发中发挥着重要作用。
2.5 配合物与BSA/HSA的相互作用
血清白蛋白(SA)是血浆中的主要蛋白质,是激素、脂类等物质的转运载体,因此研究药物−白蛋白之间的相互作用对药物的运输、释放、生物分布和毒性至关重要[41]。在本文中,研究基于芳基钌的抗癌药物和SA的相互作用对于理解芳基钌的药代动力学和药物−蛋白质相互作用非常重要。因此,我们以牛血清白蛋白(BSA)和人血清白蛋白(HSA)为模式蛋白研究了Ru-L与SA的相互作用。
BSA的结构类似于HSA,且更易获得。BSA产生荧光的生色团是由蛋白质的色氨酸(Trp)和酪氨酸(Tyr)引起的。BSA蛋白中由色氨酸的发射光谱发生变化所引起的构象转换与结合的底物或变性反应相关[42]。我们利用荧光光谱研究配合物在含有1%(V/V)DMSO 的 PBS(10 mmol·L−1,pH 7.4)溶液中与BSA(3 µmol·L−1)的荧光猝灭常数及其结合常数Ka。如图4所示,在BSA溶液中滴加配合物Ru-L(0~20µmol·L−1),可观察到BSA在334 nm处的发射峰强度急剧下降(降低率为48.68%),表明配合物与BSA有很强的相互作用。有研究表明,BSA荧光猝灭的性质可以是“静态淬灭”或“动态淬灭”[43]。静态机制通常是由猝灭剂和荧光团之间的基态结合物形成引起的,因此,它会引起荧光团吸收光谱的扰动。而在动态机制中,荧光团和猝灭剂在激发态下彼此接触,因此BSA的吸收光谱强度没有明显的变化。荧光光谱实验结果表明配合物对BSA荧光猝灭性质是“静态猝灭”,其荧光猝灭机制可以用经典Stern-Volmer[44]方程式表示:
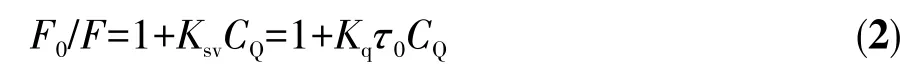
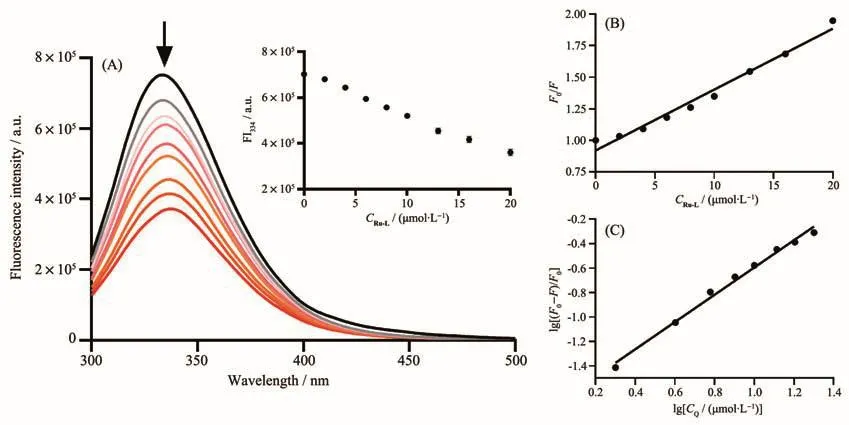
图4 配合物Ru-L在PBS中滴加BSA后的荧光光谱图:(A)BSA荧光强度随配合物浓度变化曲线,其中插图为BSA在334 nm处荧光强度对配合物浓度的拟合曲线;(B)Stern-Volmer方程拟合;(C)lg[(F0−F)/F]与 lgCQ拟合线性方程Fig.4 Fluorescence spectra of complex Ru-L after addition of BSA in PBS:(A)BSA fluorescence intensity as a function of complex concentration,where the illustration is the fitting curve of BSA fluorescence intensity at 334 nm;(B)Stern-Volmer equation fitting;(C)lg[(F0−F)/F]and lgCQfitting for the linear constants corresponding to the binding constants and complex numbers
其中F0和F分别是不存在和存在金属配合物(Q)的情况下的荧光强度;Ksv是Stern-Volmer淬灭常数,可以通过F0/F对CQ作图,由拟合直线的斜率获得。使用荧光和浓度数据的双对数曲线,可以通过截距和斜率计算得到结合常数Ka和与BSA结合的结合位点数(n):
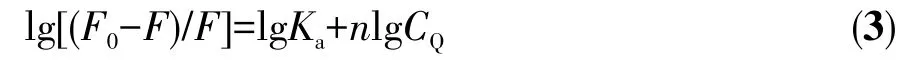
由公式(2)、(3)可算得配合物Ru-L与BSA相互作用的Stern-Volmer淬灭常数Ksv、双分子淬灭常数Kq、结合常数Ka及结合位点数n分别为 4.8×104L·mol−1、4.8×1012L·mol−1·s−1、2.0×104、1(表 2)。BSA 主要有2个Trp残基发射内源荧光,其中一个Trp位于BSA分子内部疏水腔的Trp-212位点;另一个位于分子表面的Trp-134。由于Trp-134在亲水环境会发生荧光猝灭,所以BSA分子的内源荧光主要由处于疏水腔的Trp-212产生[31]。因此,可以推断配合物Ru-L引起BSA荧光“静态猝灭”的主要原因是配合物影响Trp-212的疏水环境,从而改变BSA蛋白质的二级结构,引起荧光猝灭。
由于牛血清白蛋白(BSA)成本低且被广泛使用,在许多生化和药理学应用中可以作为人血清白蛋白(HSA)的替代品。但实际上,BSA仅具有HSA的75.8%的生物学功能,因此在许多应用中无法替代HSA[45]。HSA是585个氨基酸的球状蛋白,约占血清总蛋白的60%。它对广泛的内源性和外源性配体具有出色的结合能力,并且其丰度使其成为许多药物的药代动力学行为的重要决定因素。可根据配合物与HSA的亲和程度来研究药物进入组织的分布情况和药物代谢情况。利用荧光光谱研究配合物在含有 1%(V/V)DMSO 的 PBS(10 mmol·L−1,pH 7.4)溶液中与HSA(1µmol·L−1)的荧光猝灭常数及其结合常数Ka。如图5所示,在HSA溶液中滴加配合物Ru-L,可观察到HSA在307 nm处的发射峰强度明显下降(降低率为20.17%),表明配合物与HSA有较强的相互作用。由经典Stern-Volmer公式得到配合物Ru-L与HSA相互作用的Stern-Volmer淬灭常数Ksv、双分子淬灭常数Kq、结合常数Ka及结合位点数n分别为 4.8×104L·mol−1、4.8×1012L·mol−1·s−1、5.1×104、1(表2)。HSA中产生荧光的生色团是由蛋白质的Trp、Tyr以及丙烯氨酸(Phe)引起的,其中Trp对荧光的贡献率超过95%。研究表明,HSA中有2个主要的结构选择性结合位点,即Ⅰ位和Ⅱ位[46-48],当Ksv>103L·mol−1时,表明配合物对HSA的荧光猝灭是“静态猝灭”过程,且主要是由色氨酸及酪氨酸对HSA的微环境改变所致[49]。综上实验结果表明,配合物Ru-L改变HSA的微环境导致其荧光“静态猝灭”。
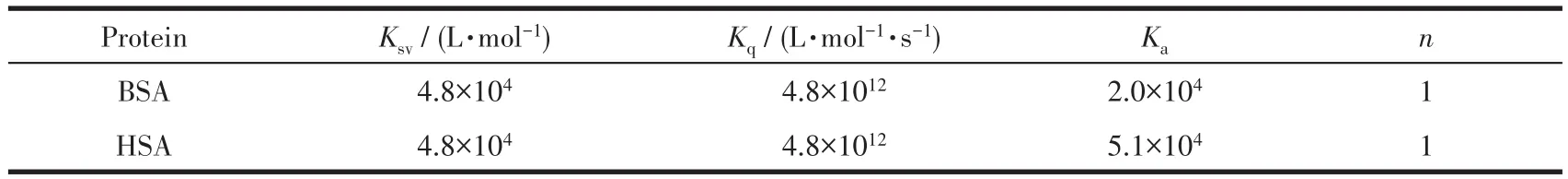
表2 配合物Ru-L对BSA/HSA的荧光猝灭数据Table 2 Fluorescence quenching data of complex Ru-L on BSA/HSA
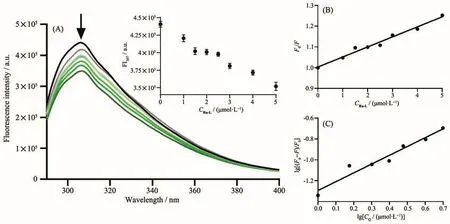
图5 配合物Ru-L在PBS中滴加HSA后的荧光光谱图:(A)HSA荧光强度随Ru-L浓度变化曲线,其中插图为HSA在307 nm处荧光强度对配合物浓度的拟合曲线;(B)Stern-Volmer方程对应双分子猝灭常数;(C)lg[(F0−F)/F]与lgCQ拟合线性方程对应结合常数及结合位点数Fig.5 Fluorescence spectra of complex Ru-L after HSA was added dropwise in PBS:(A)Curve of HSA fluorescence intensity as a function of Ru-L concentration,where the inset is a fitting curve of HSA fluorescence intensity at 307 nm versus complex concentration;(B)Stern-Volmer equation corresponding to the bimolecular quenching constant;(C)lg[(F0−F)/F]and lgCQfitting for the linear constant corresponding to the binding constant and complex number
2.6 细胞内活性氧(ROS)的检测
正常细胞通过控制适当的活性氧(ROS)水平来调控细胞内信号传导、细胞增殖及细胞死亡等生理过程[50]。有文献报道紫苏醇是通过氧化应激反应抑制了Ras/Raf/ERK通路,从而诱导细胞了死亡[51]。因此为了研究配合物Ru-L引起细胞死亡的机理,我们选择具有细胞膜穿透性的ROS荧光探针H2DCFDA检测了肿瘤细胞中ROS水平。H2DCFDA在细胞内被酯酶氧化或水解,生成无荧光的DCFH。细胞中产生的ROS可以将DCFH氧化为有荧光的DCF,因此可通过DCF的荧光强度来指示细胞内ROS的水平[52]。我们先将配合物在A2780细胞中孵育4 h,然后加入ROS荧光探针继续孵育30 min,利用流式细胞仪检测了细胞内ROS水平的变化。如图6所示,我们观察到A2780细胞中相对荧光强度仅从41.5上升到67.0,此结果相比于同类型金属配合物的ROS水平变化(大于100倍)差距较大,这表明引入紫苏醇配体的配合物Ru-L无法诱导细胞产生过量的ROS,改变了紫苏醇通过干扰氧化应激通路诱导细胞死亡的方式。
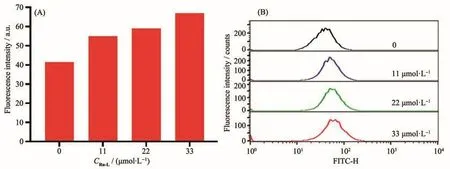
图6 A2780细胞中配合物Ru-L诱导的活性氧生成:(A)荧光强度柱状图;(B)荧光强度分布统计结果Fig.6 ROS generation induced by complex Ru-L in A2780 cells:(A)histogram of fluorescence intensity;(B)statistical results of fluorescence intensity distribution
2.7 配合物诱导A2780的细胞凋亡
配合物Ru-L不是通过干扰细胞的氧化应激通路诱导细胞死亡,因此我们利用流式细胞术进一步研究了Ru-L的作用机理。肿瘤细胞的死亡方式有细胞凋亡、自噬、坏死、焦亡及铁死亡等[53-54],其中细胞凋亡是细胞程序性死亡的过程,多数配合物通过诱导肿瘤细胞凋亡来发挥其细胞毒性。为了深入了解配合物Ru-L对肿瘤细胞毒活性的提升是否由细胞凋亡引起,我们将A2780细胞用0~33 µmol·L−1浓度的配合物孵育24 h后,通过流式细胞仪检测了细胞的凋亡效果。结果如图7所示,我们观察到配合物Ru-L诱导A2780细胞的凋亡效果以剂量依赖性的方式上升,细胞凋亡的百分数由0.597%上升到26.2%。其中配合物浓度达到1.5倍的IC50时,可诱导11.4%的A2780细胞早期凋亡及14.8%的细胞晚期凋亡。该结果表明配合物Ru-L通过诱导A2780细胞凋亡来发挥其抗肿瘤效果。
2.8 配合物诱导A2780的细胞阻滞
细胞周期主要分为间期和分裂期,其中细胞间期包括DNA复制的合成前期(G1)、DNA合成期(S)以及DNA合成后期(G2)。有文献报道紫苏醇及其衍生物诱导肿瘤细胞阻滞在G2/M期,从而诱导细胞凋亡[55]。为了深入了解配合物Ru-L对周期阻滞的影响,我们将A2780细胞用0~33 µmol·L−1浓度的配合物孵育24 h后,用流式细胞仪检测细胞的周期阻滞效果。结果如图8所示,A2780细胞的G0/G1期的细胞百分数由56.14%增加到74.08%,这就表明配合物Ru-L不同于紫苏醇对肿瘤细胞的G2/M期阻滞,而将A2780细胞周期阻滞在G0/G1期,从而诱导A2780细胞凋亡。
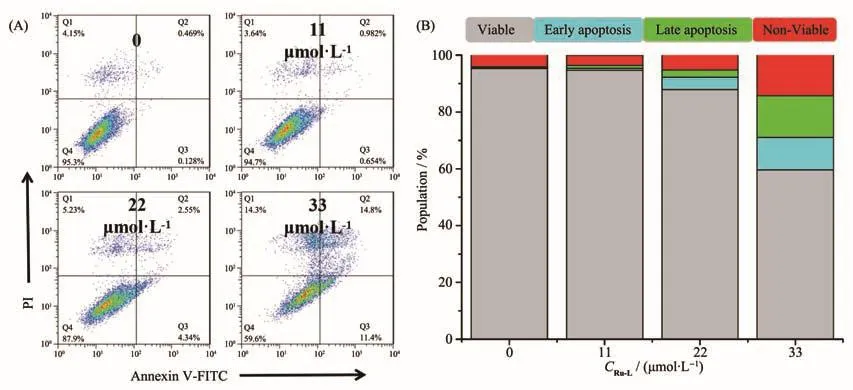
图7 配合物Ru-L对A2780细胞凋亡图:(A)细胞群落分布;(B)细胞群落柱状图统计结果Fig.7 Apoptosis of A2780 cells by complex Ru-L:(A)cell distribution;(B)histogram of cell statistical results
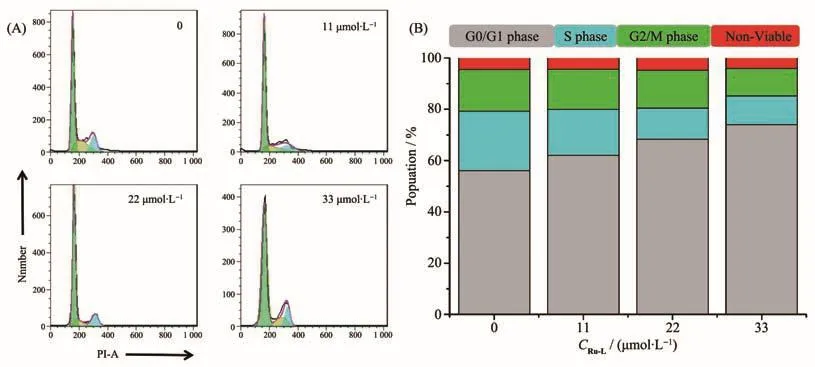
图8 配合物Ru-L对A2780细胞周期阻滞效果:(A)细胞阻滞分布;(B)细胞阻滞柱状图统计结果Fig.8 Effect of complex Ru-L on A2780 cell cycle arrest:(A)cell arrest distribution;(B)histogram of cell arrest statistical results
3 结 论
将天然产物紫苏醇进行化学修饰后引入芳基钌配合物中,得到了一种基于天然产物的芳基金属配合物Ru-L,并研究了配合物对人源肿瘤A2780、A2780/DDP及MCF-7细胞和人正常HLF细胞的抗癌活性,探究了影响芳基钌配合物抗肿瘤活性的因素。研究结果表明配合物Ru-L对肿瘤细胞表现出很高的细胞毒活性,并且对正常细胞的毒性要远低于顺铂。Ru-L具有较快的水解速度,可以嵌入DNA发生碱基堆积作用,同时还可以改变BSA/HSA的疏水环境,从而导致蛋白质的二级结构发生变化。此外,配合物通过将肿瘤细胞阻滞在G0/G1期诱导A2780细胞的凋亡,不同于紫苏醇诱导肿瘤细胞凋亡的作用机制。总之,将紫苏醇引入到芳基钌配合物中,不仅提高了紫苏醇的抗癌活性,克服了紫苏醇剂量大、毒性大的缺点,而且拓展了芳基金属配合物在抗肿瘤活性方面的应用,这些结果将为芳基金属抗肿瘤药物的研发提供一定的指导意义。
猜你喜欢
天津农学院学报(2022年2期)2022-08-05
渤海大学学报(自然科学版)(2022年1期)2022-07-25
文苑(2020年4期)2020-05-30
渤海大学学报(自然科学版)(2020年3期)2020-02-02
卷宗(2018年14期)2018-06-29
科技创新导报(2016年30期)2017-03-15
新高考·高一物理(2016年3期)2016-05-18
云南中医学院学报(2012年3期)2012-07-31