
靶向共价抑制剂的最新研究进展
2020-07-13 03:26韩王宇婧刘顺英
黄冈师范学院学报 2020年3期
韩王宇婧,刘顺英
(华东师范大学 化学与分子工程学院,上海分子治疗与新药创制工程技术研究中心,上海 200062)
共价抑制剂是一类能够与特定的靶蛋白共价结合,从而抑制其生物功能的小分子化合物。这些抑制剂往往都含有丙烯酰胺、β-内酰胺、磺酰氟基、环氧乙烷等官能团,能与靶蛋白中特定的氨基酸残基,如半胱氨酸残基、丝氨酸残基、赖氨酸残基、谷氨酸残基发生化学反应,形成共价键,从而抑制相应蛋白的生物学功能[1]。共价抑制生物靶点的小分子药物的发展历史要追溯到1897年,拜耳公司研制出了阿司匹林,用于镇痛和抗炎(图1)。青霉素抗生素代表了1940年第二次世界大战中使用的另一类经典共价抑制剂[2]。与非共价抑制剂相比较,共价抑制剂最重要的一个优点就是与靶蛋白的结合具有高亲和性,并具有相对较长的作用时效[1],因为共价化合物与靶点结合会比同类非共价药物的结合牢固得多。
共价键抑制剂通过共价键与靶蛋白相互作用的原理,成就其优点的同时,也带来了一些致命的缺点,比如因与靶点键合牢固,其一旦脱靶带来的毒副作用会比非共价抑制剂更大[2]。以至于药物发现中一度谈起共价键抑制药物,就避之不及。
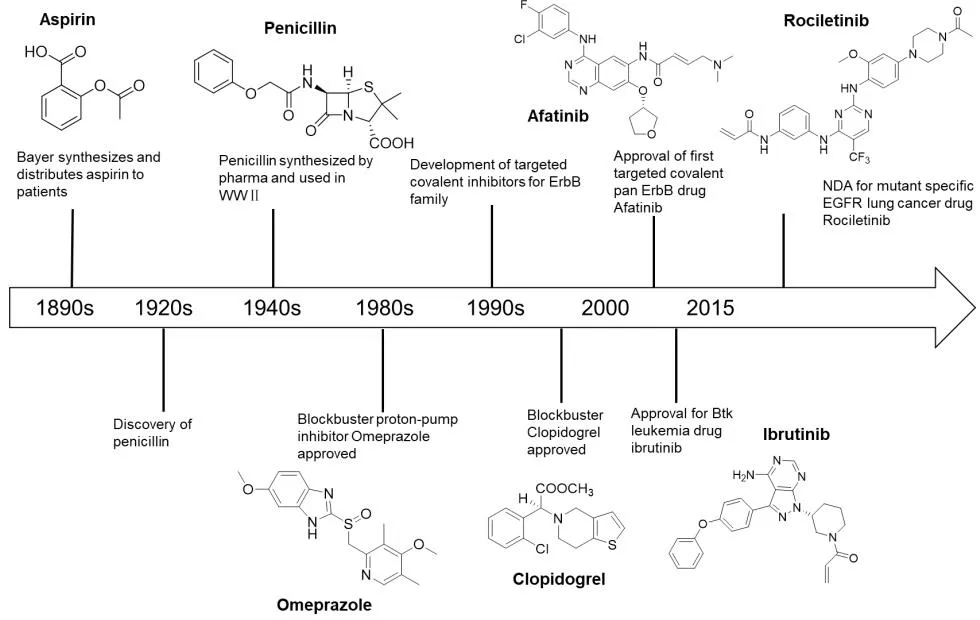
图1 靶向共价抑制剂的发展的重要事纪
但随着现代药物开发技术发展以及设计理念的更新,特别是20世纪80年代质子泵抑制剂奥美拉唑被批准上市,该药物彻底改变了与胃酸过多有关疾病的治疗方法,以及90年代抗凝血药氯格他雷和2013年非小细胞肺癌抑制剂阿法替尼的成功上市,使得人们对共价键抑制剂有了重新认识:可以通过精巧的结构设计,提高共价键与靶点氨基酸残基结合的选择性,从而克服共价键抑制剂易于脱靶的缺点,最终开发成为服务于人们健康的药物,特别是现代“基于结构理性设计药物”策略的出现,共价键抑制剂重新获得制药界的广泛关注。
此外,人们也认识到,对于一些非共价药物难以识别或者不能识别的靶点,比如药物的靶点是有着相同口袋的一类靶点家族成员,共价抑制剂也具有明显的优势。随着共价抑制剂研发热潮的回归,该领域近十余年取得了众多突破性的进展,特别是2013年第一个EGFR(表皮生长因子受体)抑制剂阿法替尼(Afatinib,1)被FDA批准上市后[3-5],已有多篇优秀的综述文章对相关研究进展进行了总结,如针对共价抑制剂作用的各种靶点,已有Renato A Bauer等人[6]进行了综述;根据靶向共价抑制剂作用的各种氨基酸残基,已有Matthias Gehringer等人[7]进行了总结;有关靶向激酶的共价抑制剂,已有Zheng Zhao等人[1]进行了综述。本文将着重介绍2013年多个共价键抑制剂获批达到共价键抑制研发的一个小高潮至今发展的一系列进入临床或作用于新靶点的靶向共价抑制剂研究进展。
目前,已报道的蛋白中能与小分子共价抑制剂发生化学反应生成共价键的氨基酸残基,主要共有以下四类: ①靶向半胱氨酸残基的共价抑制剂;②靶向丝氨酸、酪氨酸和苏氨酸残基的共价抑制剂;③靶向赖氨酸残基的共价抑制剂;④靶向谷氨酸和天冬氨酸残基的共价抑制剂。本文将按照这种分类方式对2013年以来被FDA批准或进入临床研究的代表性靶向共价抑制剂进行简要介绍。
1 靶向半胱氨酸残基的共价抑制剂
甲硫氨酸中的硫醚基团以及中性氨基酸中的巯基都只有中等的亲核性,然而一旦它在半胱氨酸侧链以硫醇盐的形式存在,其亲核性就增加了几个数量级,从而成为20种常见氨基酸中亲核性最强的亲核试剂[7]。因此,半胱氨酸是最常见的共价氨基酸残基,其侧链巯基通过亲核进攻药物分子中的亲电基团,从而形成共价键。包含缺电子基团或亲电中间体例如酮类、丙烯酰胺、炔基酰胺、α-卤代酮、炔等的化合物分子容易与半胱氨酸的巯基发生反应,形成共价键。这些亲电基团或亲电中间体常称为亲电弹头(图2)。
代表性的作用于半胱氨酸残基的小分子共价抑制剂结构见图3。阿斯利康制药有限公司研发出的奥西替尼(Osimertinib,2)于2015年被FDA批准上市,作用靶点是EGFR,用于非小细胞肺癌的治疗。前期临床研究表明,它对EGFR的L858R突变体的抑制作用可达到12nM的水平;以及对L858R/T790M突变体抑制的IC50为1nM。奥西替尼对EGFR(L858R/T790M)突变体的抑制率比野生型的大约高出200倍[8]。此后,另一个EGFR抑制剂来那替尼(Neratinib, 3)也于2018年被批准上市[9-11],用于治疗乳腺癌等疾病。口服抑制剂依鲁替尼(Ibrutinib,4)于2013年被批准上市,作为治疗B细胞恶性肿瘤药物,它对BTK(酪氨酸激酶)有着次纳摩尔的抑制活性[12-13];2017年,BTK抑制剂艾拉替尼
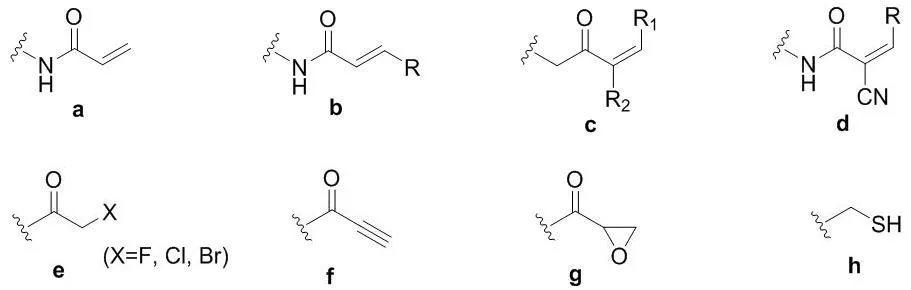
图2 作用于半胱氨酸的主要亲电弹头单元
(Acalabrutinib,5)被FDA批准上市。与第一代BTK抑制剂依鲁替尼相比,艾拉替尼具有高效及高选择性的特点[14-15]。由于它对EGFR、TEC等受体的结合能力较弱,使得它的脱靶概率很低,从而提高了它与BTK相互作用的精准性以及效率。这也大大降低了药物的副作用与毒性。Timothy Guzi课题组于2015年报道的首例选择性的FGFR(纤维原细胞生长因子受体)抑制剂BLU-554(6),现在已经进入临床研究阶段[16-17]。并于 2015 年 9 月获得了 FDA 批准的孤儿药 (orphan drug) 称号,成为第二代 FGFR抑制剂中第一个获得孤儿药批准的小分子化合物。此后,还有PRN-1371(7)、ARQ-087(8)等[18-19],也已经被批准进入临床研究。MEK1(分裂素活化的蛋白激酶1)在与癌症发生有关的信号通路中的信号转导起着关键的作用[20]。E6201(9)是一个低纳摩尔级的MEK1共价抑制剂,在临床试验中用于固体瘤和银屑病。它具有较高的血浆稳定性,从而可以用于静脉注射或者局部给药[21-22]。Cathepsin K(组织蛋白酶K)抑制剂奥当卡替(Odanacatib,10),现在正处于晚期临床研究阶段,可以治疗疏松症相关的骨质流失,从而减少老年妇女骨折的发生[23-24]。
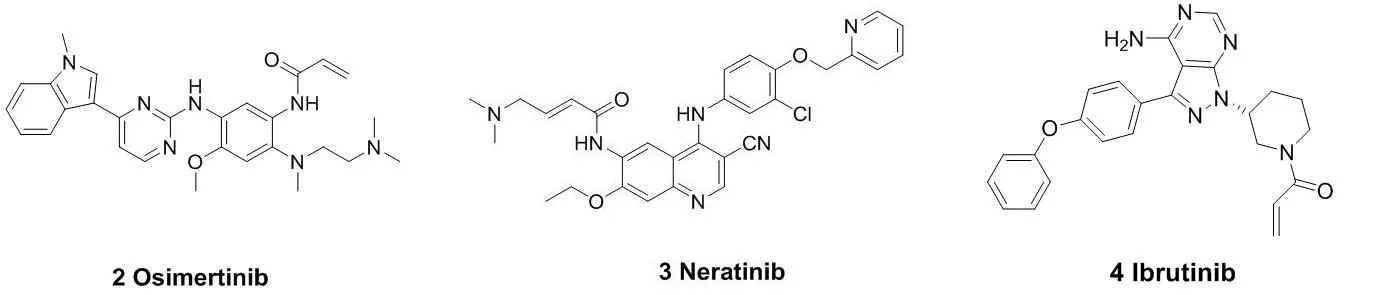
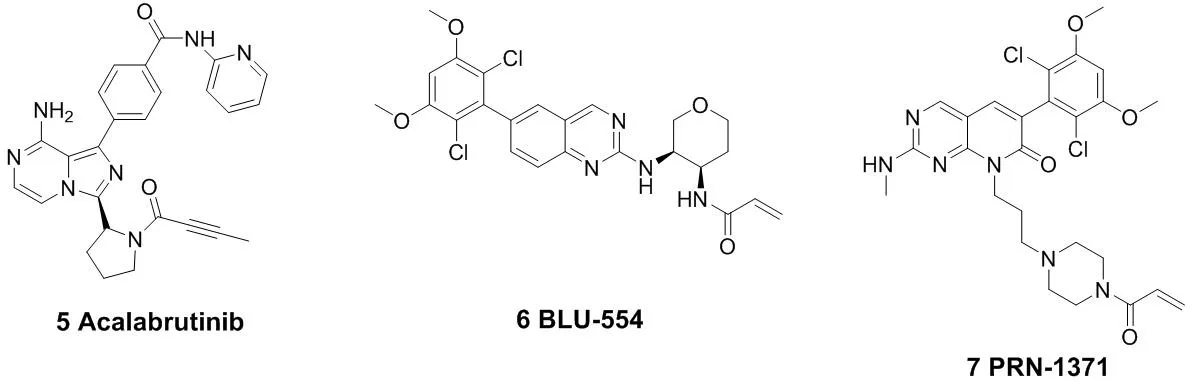
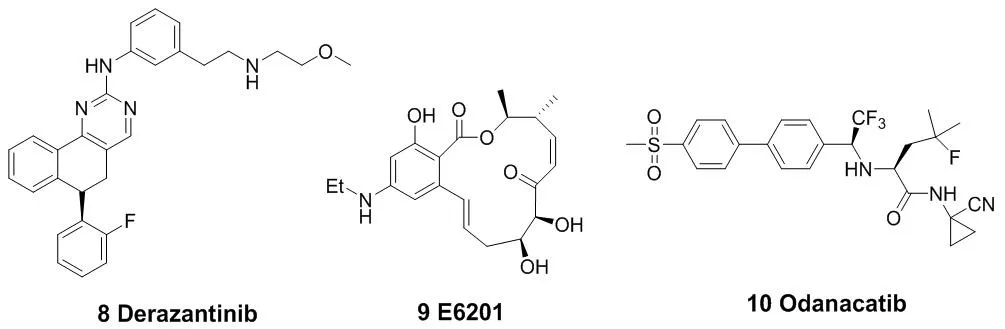
图3 代表性的作用于半胱氨酸残基的小分子共价抑制剂结构
2 靶向丝氨酸、苏氨酸和酪氨酸残基的共价抑制剂
丝氨酸、苏氨酸和酪氨酸都含有羟基,能在生化反应中与某些化合物的亲电中心发生反应从而形成共价键。
2.1 靶向丝氨酸残基的共价抑制剂
代表性的作用于丝氨酸残基的小分子共价抑制剂结构见图4。阿维巴坦(Avibactam,11)是一个作用于β-内酰胺酶的共价抑制剂,于2015年2月被FDA批准上市,用于治疗革兰氏阴性菌感染引起的相关疾病[25-26]。它不具有β-内酰胺骨架,但是能够通过共价作用使它的作用靶点β-内酰胺酶乙酰化。PF-04457845(12)是一个可以口服的FAAH(脂肪酸酰胺水解酶)共价抑制剂,现在已经进入临床研究,可以用于治疗慢性疼痛以及其他神经系统疾病[27-28]。
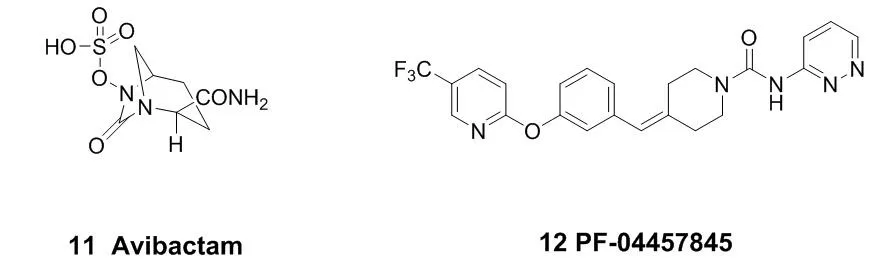
图4 代表性的作用于丝氨酸残基的小分子共价抑制剂结构
2.2 靶向苏氨酸残基的共价抑制剂
蛋白酶体是对细胞功能的调控有重要作用的一大类蛋白复合物。Salinosporamide A (13)是一个蛋白酶体抑制剂(图5),现在正处于临床三期研究阶段。它对NCI-H226非小细胞肺癌、SF-539 CNS癌、SK-MEL-28黑素瘤以及MDA-MB-435胸腺癌细胞的抑制活性均能达到10nM以下[29]。它分子结构中的β-内脂基团能够与苏氨酸残基发生反应,从而起到抑制蛋白酶体的作用[30]。
2.3 靶向酪氨酸残基的共价抑制剂
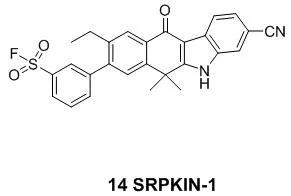
与半胱氨酸或者未质子化的赖氨酸相比,中性的酪氨酸残基的固有亲核性相对较弱,因此,开发出作用于酪氨酸残基的共价抑制剂是相当具有挑战性的,这也导致以酪氨酸残基为作用靶点的共价药物较少[7]。2018年4月哈佛大学报道了首例作用于SRPK1/2(丝氨酸蛋白激酶)的不可逆抑制剂SRPKIN-1(14)(图6)[31]。在ATP键合口袋中,它能够与酪氨酸的酚羟基形成共价键从而抑制丝氨酸蛋白激酶的活性。
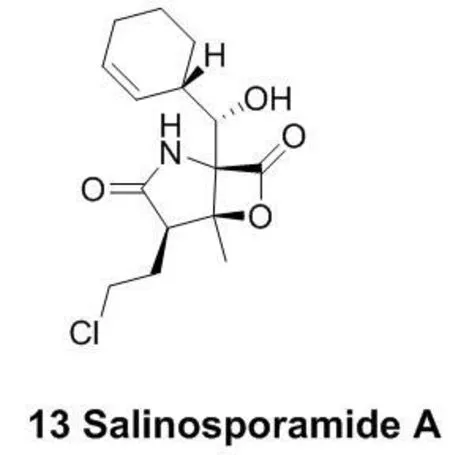
图5 代表性的作用于苏氨酸残基的共价抵制剂结构
3 靶向赖氨酸残基的共价抑制剂
赖氨酸中亲核性的氨基能与不可逆抑制剂作用。由于氨基在蛋白界面的pKa值大约为10.4,而生理环境的pH值大约7.4,因此在生理条件下大部分赖氨酸残基都以质子化的形式存在,从而无法作为亲核试剂。这对它在生物体中的反应带来了一些挑战。磷脂酰肌醇激酶(PI3K)是促进磷脂酰肌醇的肌醇环磷酸化的信号转导酶家族中的一员,它在细胞增殖、凋亡以及细胞其他功能中起着重要的作用[6]。化合物18的结构以及其与谷氨酸残基结合的作用机制(图7)。天然药物渥曼青霉素(Wortmannin,15)就是一个PI3K抑制剂[32],但是其稳定性及选择性较差。考虑到Wortmannin在与赖氨酸残基结合时分子中的呋喃环打开,故将呋喃环用开链的胺替代,一个稳定的口服PI3K抑制剂PX-866(16)被开发出来,现在已进入临床研究阶段。PX-866能与PI3K催化位点上的赖氨酸残基发生一个乙烯基转酰胺化反应,从而不可逆的抑制PI3K激酶[33-34]。2015年纽卡斯特大学报道了首例CDK2(周期蛋白依赖性激酶)共价抑制剂NU6300(17),可用于治疗具有确定遗传特征的肿瘤[35]。此外,蛋白质激酶的不可逆共价抑制剂大多数是以半胱氨酸的巯基作为亲核基团来开发的, 而基于赖氨酸的氨基来开发的却较为少见,NU6300的开发也对科学家们开发具有类似弹头单元的药物具有重要启示。
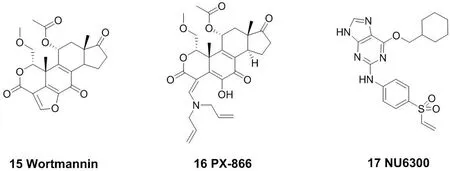
图7 代表性的作用于赖氨酸残基的小分子共价抑制剂
4 靶向谷氨酸和天冬氨酸残基的共价抑制剂
谷氨酸和天冬氨酸都含有羧基,羧基的弱亲核性给共价修饰谷氨酸和天冬氨酸侧链带来了一定的困难[6],这里仅介绍一例作用于谷氨酸羧基的共价抑制剂。PDEδ是磷酸二酯酶中的一个δ亚型,在维持细胞中KRAS的动力学分布中起着不可替代的作用[36]。
马普分子生理学研究所(Max Planck Institute of Molecular Physiology)的Herbert Waldmann课题组,在2017年报道了一个作用于PDE的δ亚型上的谷氨酸残基的共价抑制剂化合物18[37]。化合物18分子中的N-甲基异恶唑盐能在碱的作用下开环,形成乙烯酮酰亚胺,从而与羧基反应形成共价键[38],作用机制如图8。
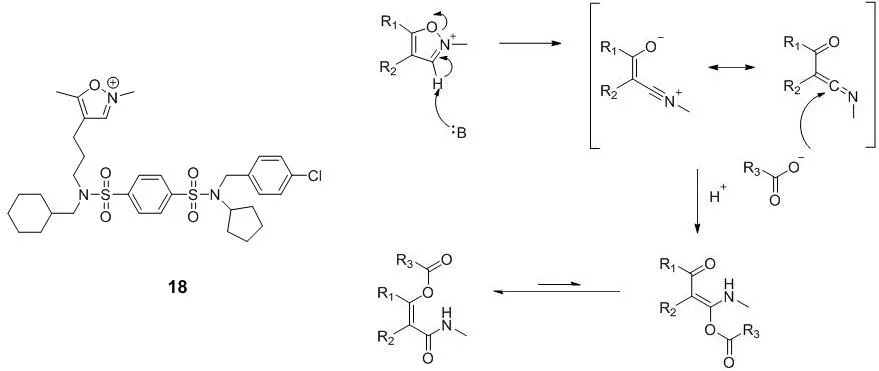
图8 化合物18的结构以及其与谷氨酸残基结合的作用机制
本文简单介绍了2013年小分子共价键抑制剂获批为药物的小高潮以来,共价抑制剂的发展历史和一些有代表性的共价药物及他们与靶点的作用机制。靶向共价抑制剂的设计是一个有前景的领域,且正处于茁壮成长的阶段。与非共价抑制剂相比,共价抑制剂对靶点的作用更高效,从而能够通过减少给药剂量来提高治疗窗口。此外,共价键抑制剂在减少靶点突变导致的耐药性(drug resistance)和药物开发过程中不需要PK药代动力学研究等优势,近年来在制药界得到了重新认识和重视。但如何开发高选择性的共价键抑制剂以避免脱靶效应带来的毒副作用仍然是这一领域研究的关键问题。随着现代基于结构的理性药物设计理念以及化学生物学等新兴学科、技术的发展,以及对靶点蛋白结构、性质更深入的认识,这一问题必将在不远的将来得到有效解决。除了本文中提到的一些有代表性的靶向共价抑制剂之外,大量各种各样的共价抑制剂正在不断涌现;靶向共价抑制剂的开发技术也在飞速发展。我们有理由相信,靶向共价抑制剂开发必将迎来新的研发高潮,造福人类健康。
猜你喜欢
中国农业科学(2022年12期)2022-06-28
药学进展(2022年1期)2022-03-06
中国畜牧杂志(2022年1期)2022-01-20
中学生理科应试(2021年10期)2021-12-07
南昌大学学报(医学版)(2021年1期)2021-11-29
潍坊学院学报(2021年2期)2021-07-22
食品与生物技术学报(2021年2期)2021-01-16
合成树脂及塑料(2021年6期)2021-01-09
中学生数理化(高中版.高二数学)(2020年6期)2020-07-24
中学化学(2015年8期)2015-12-29
