
脂联素通过Nrf2/HO-1通路对急性心肌梗死大鼠心功能和心肌细胞凋亡作用的机制研究
2020-07-12 11:59李媛莉赵继波张三明
中国循证心血管医学杂志 2020年5期
李媛莉,赵继波,张三明
急性心肌梗死(AMI)后的氧化应激反应和细胞凋亡是心功能损伤的主要机制,改善氧化应激反应和抑制急性心肌缺血后的细胞凋亡是治疗AMI的主要策略[1]。核因子E2相关因子2(Nrf2)/血红素加氧酶-1(HO-1)通路是调节氧化应激的关键通路,在AMI继发损伤中发挥重要作用[2],研究显示激活Nrf2/HO-1通路可缓解缺血再灌注后心肌损伤[3]。脂联素(APN)是一种由脂肪细胞分泌的细胞因子,具有调节新陈代谢、保护血管和心脏的作用。最新研究显示在心肌缺血再灌注损伤大鼠模型中,APN的水平显著降低,且APN水平与心肌梗死面积呈负相关[4],提示其具有保护AMI心肌损伤的作用。本文分析APN对AMI大鼠心功能的影响,并探究其对Nrf2/HO-1影响的保护机制。
1 材料与方法
1.1 动物、试剂和材料选取45只SPF级雄性SD大鼠220~250 g(南京金陵医院,中国),APN(Sigma公司,美国),Mayer's苏木精和0.5%曙红水溶液(H8070,DH0050,北京Solarbio公司,中国),酶联免疫吸附(ELISA)试剂盒、TUNEL凋亡试剂盒(碧云天公司,中国),酶标仪(Model 680,Bio-Rad,美国),RIPA裂解缓冲液(中国,北京,Beyotime),BCA蛋白测定试剂盒(北京Applygen公司,中国),抗体购自美国Abcam公司,PVDF膜(Bio-Rad公司,美国)。小动物超声成像仪(麟得科学仪器有限公司,中国),BX-42光学显微镜(Olympus Corporation,日本)。
1.2 动物分组、建模和干预45只大鼠随机分为3组:Sham组、AMI组和AMI+APN组,每组各15只。AMI组和AMI+APN组大鼠通过结扎方法建立AMI模型[5],首先腹腔注射1%戊巴比妥(剂量为3 ml/kg)麻醉大鼠,麻醉后连接心电图,剪开胸腔暴露心脏,使用6/0线在左心耳下约2 mm处的冠状动脉前降支结扎。结扎后心电图显示大鼠心电图显示QRS波增宽,则为结扎成功,保持结扎30 min后,取出丝线进行再灌注,缝合胸腔。术后注射8万单位的青霉素抗感染干预,持续3 d。Sham组打开胸腔暴露心脏但不结扎,缝合胸腔后注射青霉素,方法与AMI组相同。AMI+APN组大鼠在建模第2 d使用APN腹腔注射,剂量为10 μg/kg,Sham组和AMI组给予等量生理盐水干预1/d,连续7 d。
1.3 观察指标及方法
1.3.1 心功能指标通过超声成像仪行心脏功能检查,检测指标包括左室射血分数(LVEF)和左室内压力最大/小变化率(±dp/dt max)。
1.3.2 心肌酶指标大鼠麻醉后处死,收集血液样本,在2000 rpm离心20 min后,取上层血清。应用ELISA试剂盒检测肌酸激酶(CK-MB)和乳酸脱氢酶(LDH),根据说明书加入抗体,通过酶标仪检测吸光度,根据标准曲线计算浓度。
1.3.3 HE染色大鼠处死后取出心脏,并将心脏组织在室温下用4%多聚甲醛中固定过夜,室温下通过梯度浓度的乙醇处理(80%持续2 h,90%持续2 h,95%过夜,100%分别持续0.5 h、0.5 h和1.0 h),然后包埋在石蜡中。将石蜡样品切成4 μm厚的切片,Mayer's苏木室温下精染色10 min,0.5%曙红水溶液室温下染色3 min。细胞核和其他酸性结构被染成蓝色,而细胞质被染成红色。使用光学显微镜以×400放大率采集图像。
1.3.4 TUNEL染色检测心肌细胞凋亡心肌组织按照1.3.3的方法将组织固定并切成10 μm的厚度切片,使用DAPI染色细胞核,加入TUNEL试剂染色凋亡细胞。在显微镜×400放大倍数下随机选择五个视野,蓝色荧光为细胞核,呈现绿色荧光的细胞计为凋亡细胞,凋亡率=(绿色荧光细胞数目/视野中细胞核总数目)×100%。
1.3.5 Western blot在液氮下将心肌组织研磨并使用RIPA裂解缓冲液裂解,用BCA蛋白测定试剂盒测量总蛋白含量。SDS-PAGE分离等量的总蛋白(120 V下电泳90 min),将蛋白转移到PVDF膜上(50 V,120 min),在含5%脱脂牛奶的封闭溶液中封闭。将PDVF膜与anti-Nrf2、anti-OH-1(1:500稀释)4°C孵育过夜,加入1:5000稀释的HRP偶联二抗在室温下孵育1 h。应用ECL试剂盒可视化蛋白条带,ImagePD软件使用GAPDH作为内参对蛋白条带的灰度进行定量。
1.4 统计学处理所有实验设立3个复孔作为平行实验。数据以平均值±标准偏差(SD)表示。统计分析使用SPSS 19.0软件。3组间比较进行单因素方差分析,两两比较使用SNK-q检验。统计学显着性表示为P<0.05。
2 结果
2.1 APN对AMI大鼠心功能的影响AMI组大鼠LVEF(44.24±6.58%)、±dp/dt max(3.84±1.16×103mmHg/s,2.81±0.90×103mmHg/s)水平显著低于Sham组(P<0.05),AMI+APN组LVEF(61.46±6.54%)、±dp/dt max(4.89±1.45×103mmHg/s,3.52±1.33×103mmHg/s)水平显著高于AMI组(P<0.05)(表1)。

表1 各组大鼠LVEF和±dp/dt max比较
2.2 APN对AMI大鼠心肌酶指标的影响AMI组CK-MB(109.68±9.65 kU/L)和LDH(134.26±11.92 kU/L)显著高于Sham组(P<0.05),AMI+APN组CK-MB(71.69±8.74 kU/L)和LDH(97.56±12.44 kU/L)显著低于AMI组(P<0.05)(表2)。
2.3 心肌HE染色结果Sham组心肌细胞形态呈梭形,且细胞与细胞之间排解紧密。AMI组心肌细胞形态异常,细胞间出现大量空隙并排列紊乱。AMI+APN组心肌细胞排列基本正常,空隙显著减少(图1)。

表2 各组CK-MB和LDH水平比较

图1 HE染色检测心肌组织损伤情况(×400)
2.4 各组心肌细胞凋亡情况比较蓝色荧光为细胞核,绿色荧光为凋亡细胞。AMI组的凋亡率(44.21±3.98%)显著高于Sham组(3.18±0.08)(P<0.05),AMI+APN组心肌细胞凋亡率(23.58±3.78%)显著低于AMI组(P<0.05)(图2)。
2.5 各组Nrf2/HO-1通路水平比较与Sham组比较,AMI组Nrf2和HO-1水平显著降低(P<0.05),AMI+APN组Nrf2和HO-1水平显著高于AMI组(P<0.05),(表3、图3)。
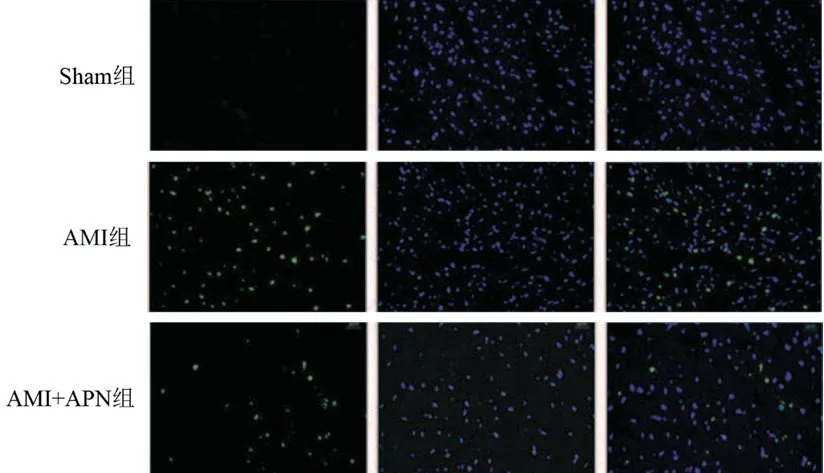
图2 TUNEL染色检测心肌细胞凋亡情况(×400)

表3 各组Nrf2和HO-1蛋白水平比较
3 讨论
AMI是世界范围内引起人类死亡的主要原因之一,心肌组织的血液灌注受阻会引起氧自由基等活性氧物质的产生增加,促进有害细胞信号的激活,引起血管内皮细胞或心肌细胞的炎性细胞因子和趋化因子的表达,导致炎性反应并引起心肌损伤,缓解氧化应激反应是治疗AMI的重要途径[6]。
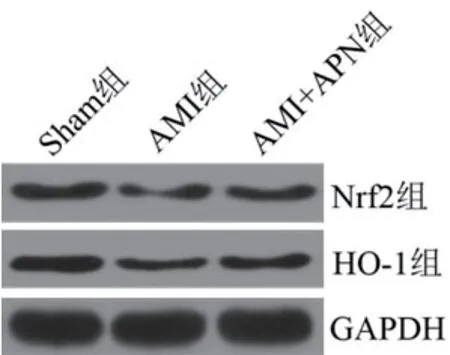
图3 Western blot检测Nrf2/HO-1通路中蛋白水平
APN是一种由脂肪细胞和心肌细胞分泌的细胞因子,有调节线粒体的作用,APN降低会引起线粒体损伤和氧化应激反应。最新研究发现,APN与ST段抬高型AMI患者病情有关,病变越重的患者APN水平越低[7]。徐小静等[8]研究也显示了APN与缺血引起的组织损伤有关。有体内实验显示APN干预可缓解2型糖尿病大鼠的氧化应激水平[9]。据此我们推测APN可能会缓解AMI引起的心肌损伤。本文通过结扎方法建立AMI大鼠模型,并通过心电图证实建模成功,用腹腔注射APN干预。结果显示应用APN干预后,大鼠心肌损伤情况显著缓解,并降低了心肌酶指标,具有改善心功能的作用。通过本研究,我们发现APN干预可有效缓解AMI引起的心肌损伤并保护心功能。
为进一步探究APN缓解AMI心肌损伤的机制,我们对心肌细胞的凋亡情况进行检测。梗死区域以及后续心肌细胞的损伤和凋亡是引起AMI患者心肌损伤和心功能受损的重要病理基础,这会导致左心室重塑、纤维化甚至心力衰竭,抑制梗死后心肌细胞凋亡是保护心肌功能的重要途径[10]。本实验显示,AMI组凋亡率(44.21±3.98%)显著高于Sham组(3.18±0.08),AMI+APN组心肌细胞凋亡率(23.58±3.78%)显著低于AMI组,说明APN可显著缓解AMI引起的心肌细胞凋亡。Nrf2/HO-1通路是抗氧化的重要途径,研究显示Nrf2和HO-1水平上调可缓解缺血引起的氧化应激损伤[11],且Nrf2/HO-1通过调节氧化应激或炎性反应的方式,直接调控心肌细胞的凋亡[12,13]。本研究还显示AMI组Nrf2和HO-1水平显著低于Sham组,AMI+APN组Nrf2和HO-1水平显著高于AMI组。有研究显示APN可通过激活Nrf2相关通路改善高血糖引起的心脏肥大和功能障碍[14]。Enji等[15]研究显示APN具有激活Nrf2通路的作用。这提示在AMI大鼠模型中,APN可能通过激活Nrf2/HO-1通路抑制心肌细胞凋亡。
综上所述,APN可能通过促进Nrf2/HO-1通路抑制AMI模型大鼠的心肌细胞凋亡,从而发挥保护心功能的作用。但关于APN对AMI的影响及调控Nrf2/HO-1通路的机制仍需进一步研究。
猜你喜欢
世界科学技术-中医药现代化(2022年2期)2022-05-25
昆明医科大学学报(2022年4期)2022-05-23
上海交通大学学报(医学版)(2022年3期)2022-05-05
湖南畜牧兽医(2021年6期)2022-01-24
食品安全导刊(2021年21期)2021-08-30
猪业科学(2021年5期)2021-06-02
中国畜禽种业(2021年4期)2021-05-21
心肺血管病杂志(2020年5期)2021-01-14
世界科学技术-中医药现代化(2020年2期)2020-07-25
科学咨询(2020年10期)2020-04-01
