
苏氨酸不同分子构型太赫兹吸收峰的量子化学指认
2020-07-08 14:30王志春刘成毫
光谱学与光谱分析 2020年7期
李 伟, 燕 芳, 王志春, 刘成毫
内蒙古科技大学信息工程学院, 内蒙古 包头 014010
引 言
太赫兹(THz)辐射指波长在30 μm~3 mm之间的电磁波。 太赫兹波段不仅是高效的信息载体, 且太赫兹波光子能量极低, 能穿透非金属和非极性材料, 因此在对物体内部缺陷的探测和对隐藏物的检测时具有很大的优势。 目前, 太赫兹光谱技术在无损检测技术[1]、 生物医学研究、 生物化学研究、 安全检查、 军事应用[2-3]等领域都展现出巨大的潜力。 许多生物分子的集体振转模式均位于太赫兹波段, 故不同生物分子在太赫兹吸收谱中呈现出不同的吸收峰, 即生物分子在太赫兹波段具有“指纹性”, 获得待测物质的太赫兹吸收谱后, 与标准指纹谱库进行对照可以实现对待测物质的定性分析。
同时含有氨基和羧基的有机化合物统称为氨基酸, 是构成蛋白质大分子的基础结构, 几乎一切生命活动都与之相关。 因太赫兹波可以穿透氨基酸分子, 与可见光和红外光谱技术相比, 太赫兹散射较小, 并且不存在有害光致电离, 与紫外光谱技术相比, 不会引起待测样品变性, 能够安全无损地检测氨基酸样品。 因此作为红外、 紫外等光谱技术重要补充手段的太赫兹光谱技术可为氨基酸材料鉴别提供新方法。
1 实验部分
采用透射式太赫兹时域光谱系统进行苏氨酸样品实验谱的测定。 图1为本实验用透射式太赫兹时域光谱系统原理图, 光谱仪及飞秒激光器等主要装置的性能参数如表1所示。
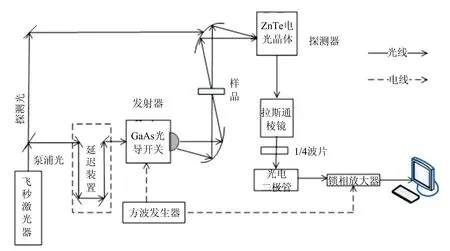
图1 透射式太赫兹时域光谱系统原理图

表1 太赫兹时域光谱系统主要装置的性能参数
为了减少空气中的水分对太赫兹波的吸收, 实验前在太赫兹光路中充入干燥的高纯度氮气, 确保光路密闭系统在实验时保持在室温25 ℃, 湿度控制在4%以下。
在实验过程中太赫兹波作用简图如图2所示, 太赫兹波垂直打在样片上, 其中黑色部分为样片。 对参考波形和待测样品波形做傅立叶变换, 参考光谱和信号光谱依次是
Eref(ω)≡Ar(ω)exp[-iφr(ω)]
(1)
Esam(ω)≡As(ω)exp[-iφs(ω)]
(2)
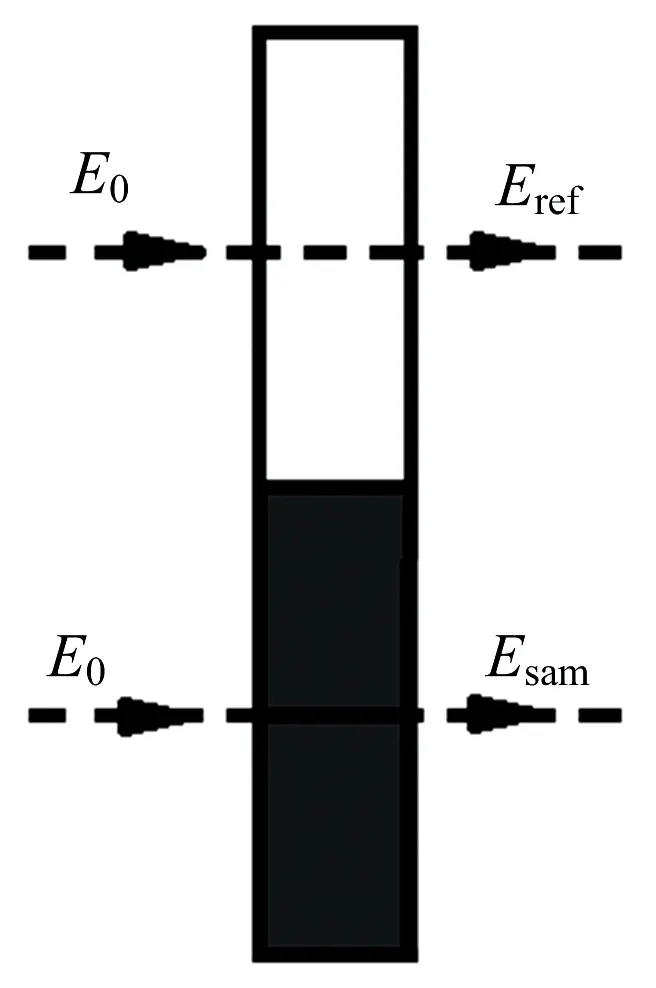
图2 太赫兹波通过样片和空气的模型图
式(1)和式(2)中A(ω)为太赫兹辐射的幅值,φ(ω)为太赫兹辐射的相位。
取Eref(ω)和Esam(ω)比值的幅频特性和相频特性[见式(3)]
(3)
(4)
折射率和吸收系数等光学常数是表明待测样品宏观光学性质的基本物理量。 由式(4)得折射率[见式(5)]
(5)
由折射率关于频率的波形得样品的吸收系数
(6)
式(6)中,α(ω)为吸收系数。
为了降低颗粒度差异引起的太赫兹波散射, 实验所用进口苏氨酸样品经充分研磨后加入聚乙烯粉末混合, 聚乙烯粉末颗粒直径约为53~75 μm, 购买于美国Sigma-Aldrich公司, 以10 MPa压力冲压成片, 表2为样品配比表。
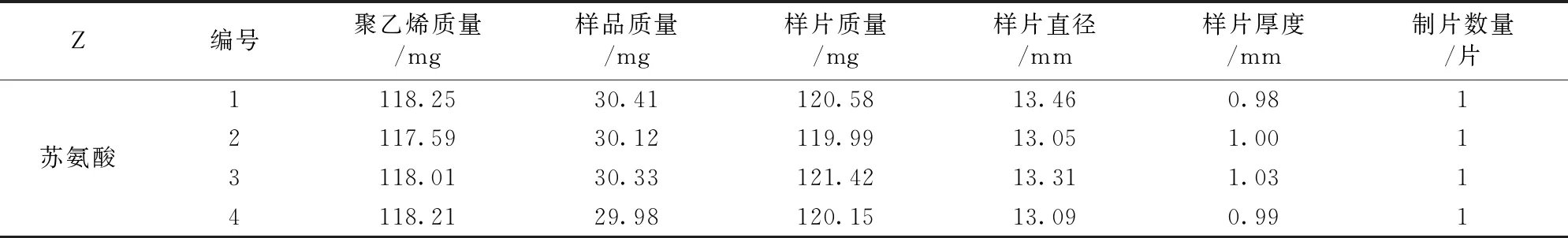
表2 样品配比表(湿度<4%, 温度25 ℃)
实验中, 为了减小太赫兹时域光谱系统测量样品信号和参考信号的误差, 每组样品测量三次后取其平均值, 由式(6)得到进口苏氨酸样品的太赫兹吸收谱如图3所示。
2 量子化学模拟计算
量子化学建模就是绘制分子结构, 分子构型可以在GaussView中直接绘制, 也可以在ChemDraw中通过构建分子式的方法转出分子构型。 利用GaussView绘制分子构型需有原子坐标、 键长、 键角等先验知识, 而ChemDraw转出的分子构型直接具备原子坐标、 键长、 键角等信息。
结构优化在红外光谱、 振动、 偶极矩等分析中格外重要, 优化的目的是将绘制或编写的待计算结构的原子大小、 键长、 键角等更合理, 这种“合理”的标准实际上是要找到体系总能量最低的一种符合客观实际的待计算化学结构。
Gaussian03和Materials Studio示出的红外吸收光谱为波数-能量(nλ-E)数据, 波数(nλ)为波长的倒数, 为了使吸收谱曲线更为直观, 通常将横轴的波数转换为频率, 波长与频率之间的关系为
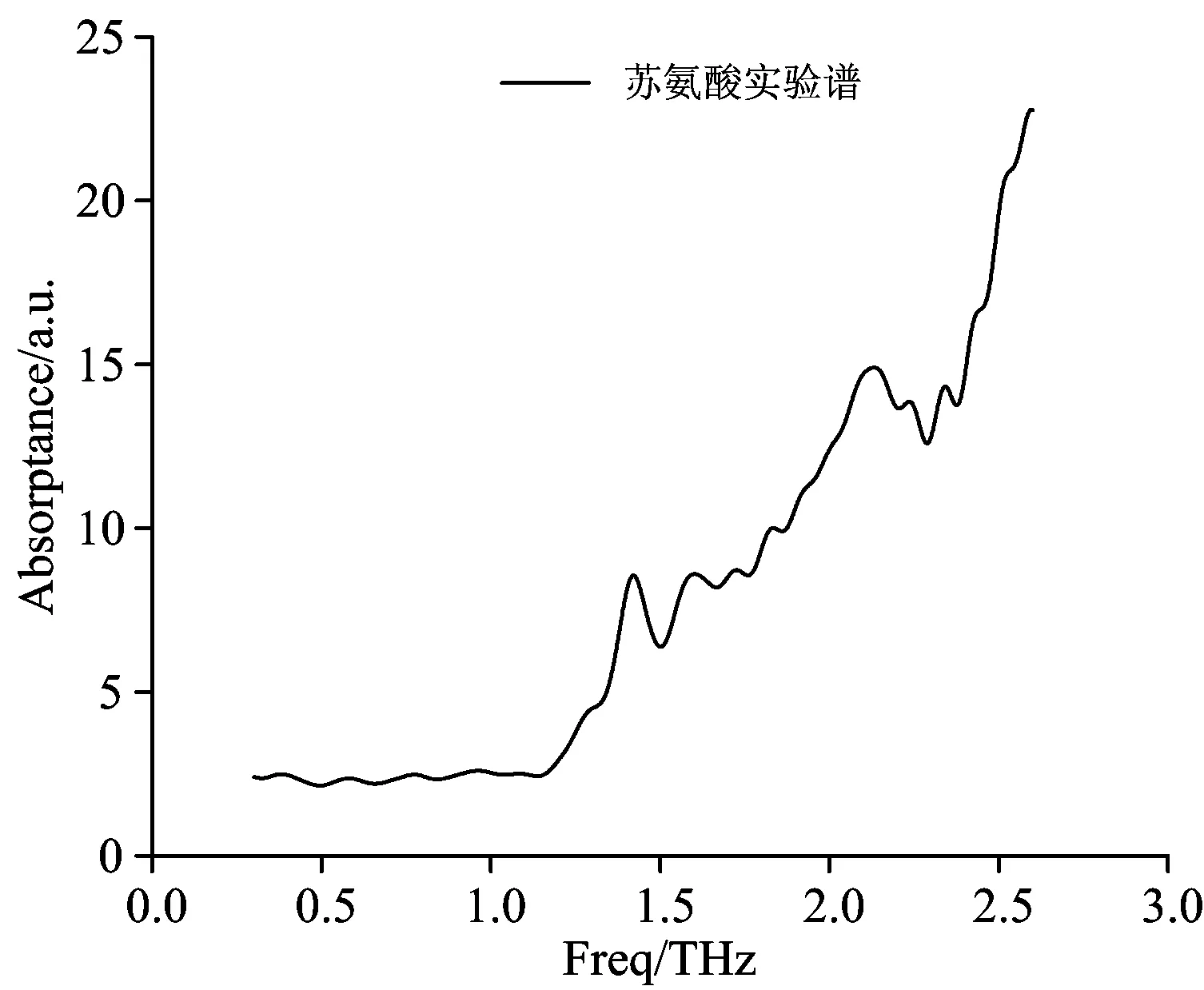
图3 苏氨酸样品的太赫兹吸收谱
(7)
式(7)中, 取真空光速c=2.998×108m·s-1, 则频率为
f=0.029 98×nλ
(8)
氨基酸在固体样品中以两性离子形式存在, 为了模拟得到与实际样品最为吻合的结果, 对苏氨酸单分子的两性离子进行计算分析。 分子构型在ChemDraw中绘制, 导出分子结构的笛卡尔坐标并导入Gaussian03进行结构优化和振动光谱计算: 因两性离子带正负两性电荷, 而选用6-311G++(d, p)基组, 同时采用基于B3LYP杂化泛函的密度泛函理论做构型优化, 最后进行红外振动及光谱(IR)计算。 由于太赫兹波处于远红外波段, 故太赫兹吸收光谱需在红外吸收光谱的远红外段提取。 如图4所示为苏氨酸两性离子形式存在的单分子构型, 右上角为两性离子形式的苏氨酸单分子结构式。
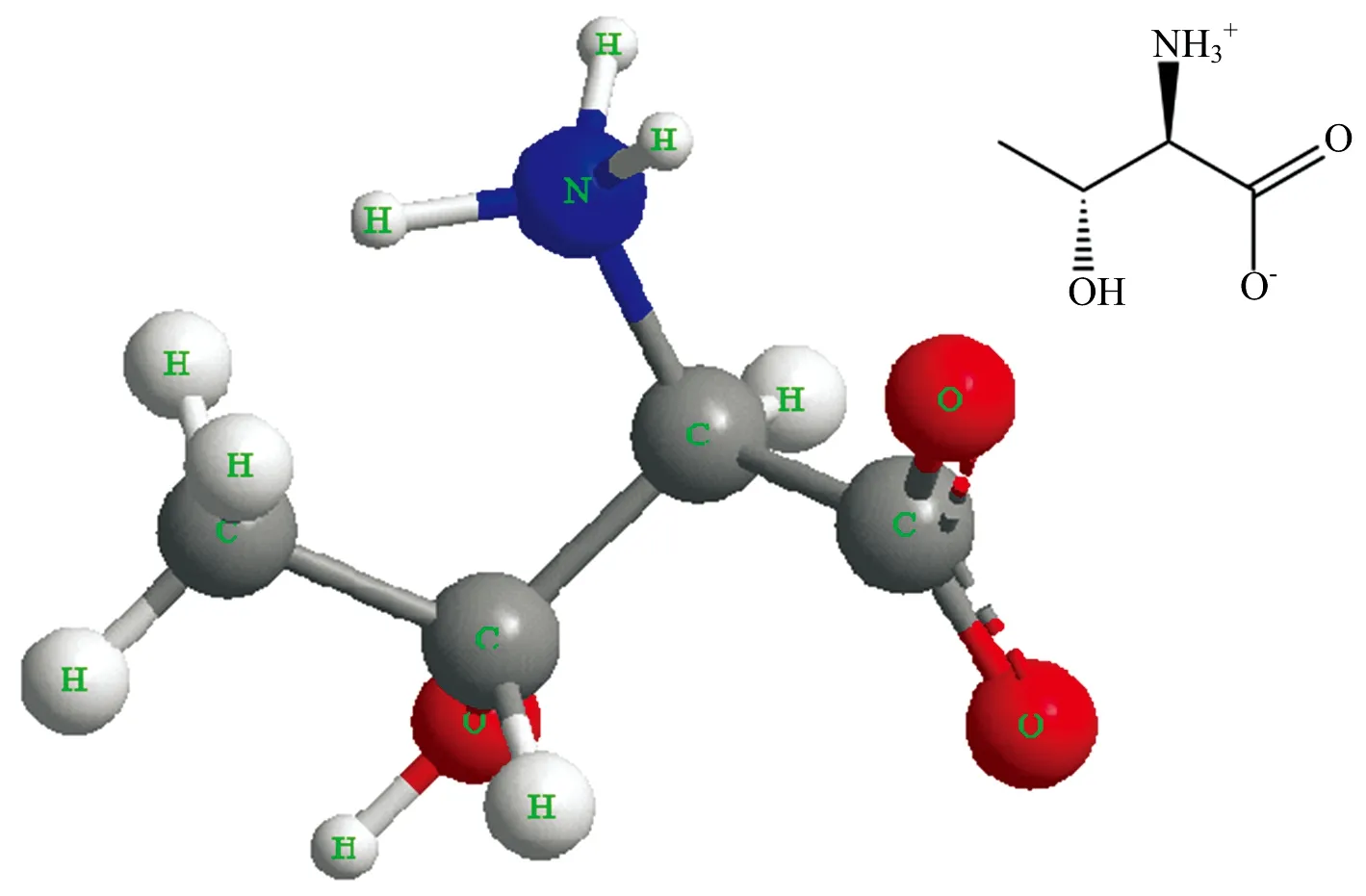
图4 苏氨酸样品的单分子构型
聚合体理论指出: 带强电正性粒子(如N原子和N3+离子)易与带强电负性粒子(如O原子和O2-离子)通过氢键作用形成聚合体。 在苏氨酸中, 羧基中氢氧根离子(OH-)的氧原子带一个单位强电负性, 而氨基中的氮原子带一个单位强电正性, 故在氮氧之间易形成不稳定化学键——氢键, 使苏氨酸的氨基酸官能团形成聚合体内环。 特别是, 在n个苏氨酸分子中能形成n个氢键, 并且m个苏氨酸分子形成的聚合体称为m聚苏氨酸。 二聚体为苏氨酸聚合体的最小构型, 计算二聚体构型的太赫兹吸收谱, 能考虑分子间氢键作用对太赫兹的影响。 图5为苏氨酸二聚体构型, 右上角为两性离子形式苏氨酸二聚体结构式。
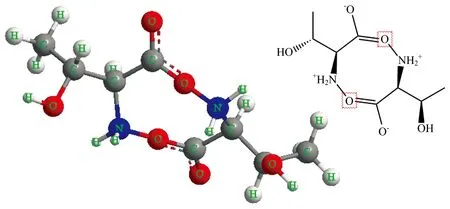
图5 苏氨酸二聚体构型
一些物质保持其物理性质的是晶胞, 晶胞由两个及两个以上构成该物质的同种分子构成。 苏氨酸的物理性质由晶胞保持。 通过英国剑桥晶胞数据库(CCDC)查得苏氨酸晶格参数为a=13.630 Å,b=7.753 Å,c=5.162 Å,V=545.486 Å3, 晶胞需要在Materials Studio中的CASETUP模块中运算, CASTEUP是基于固相平面波赝势密度泛函的计算方法。 苏氨酸晶胞构型的太赫兹声子振动选取平面波赝势截断能750 eV进行结构优化, 结果存在过度优化, 进而选择GGD基组对PBE方法的改进方法PBEsol进行再优化, 然后进行声子振动分析得到苏氨酸晶胞的太赫兹吸收谱。 图6为苏氨酸晶胞结构, 从图中可以看出, 保持苏氨酸的物理性质最少需要四个苏氨酸分子。

图6 苏氨酸晶胞结构
上述苏氨酸单分子、 二聚体、 晶胞构型的量子化学计算均在配置有数据处理器2.6 GHz主频, 单核四线程, 缓存数据存储器128G固态硬盘(SSD)的计算机中。 计算机时与收敛迭代次数限于原子个数、 分子空间结构和数据处理器配置等, 表3为各结构的计算机时和收敛迭代次数。

表3 计算机时及收敛迭代次数
3 结果与讨论
图7为苏氨酸单分子构型、 二聚体构型计算谱与实验谱的对比, 图中实线为实验谱, 虚线为单分子构型计算谱, 点划线为二聚体构型计算谱。
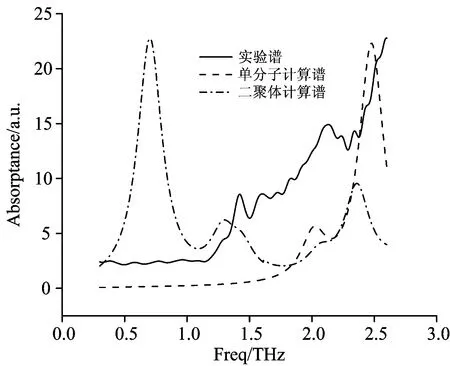
图7 苏氨酸实验谱与单分子、 二聚体计算谱的对比
Fig.7 Comparison of threonine experimental spectrum with single molecule and dimer calculated spectrum
结果显示, 考虑了分子间氢键作用的二聚体构型的太赫兹吸收谱已具备与实验谱一致的吸收峰个数, 但除2.2~2.6 THz段外的峰位均有较大红移, 同时与单分子构型计算谱相比, 单分子构型计算谱的基线较好。 为了研究能否获得吸收峰个数和峰位均基本一致的实验谱与计算谱, 需要考虑分子间范德华力对太赫兹谱的影响。
图8为苏氨酸晶胞构型计算谱与实验谱的对比, 图中实线为实验谱, 虚线为计算谱。

图8 苏氨酸晶胞构型计算谱与实验谱的对比
结果显示, 苏氨酸晶胞构型计算谱无论是吸收峰个数还是峰位都与实验谱基本吻合, 晶胞构型兼顾了分子间氢键作用与范德华力作用, 在太赫兹辐射作用下也与实际样品结构较为吻合。 表4列出了晶胞构型计算谱与实验谱的对比, 计算谱与实验谱的相对误差均小于10%(0.26 THz), “-”号表示计算谱结果较实验谱红移。
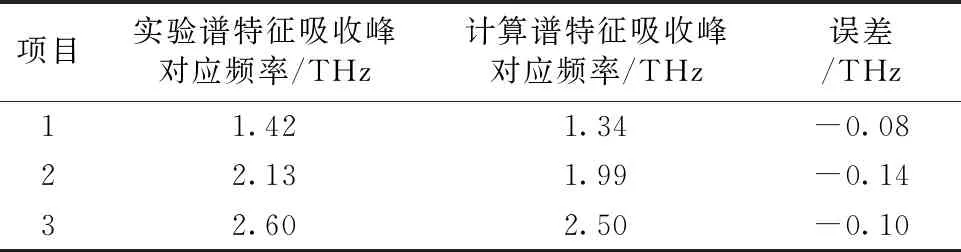
表4 晶胞构型实验谱与计算谱的对比
值得指明, 计算谱吸收峰峰位存在明显红移, 造成差异的原因归结为以下两点:
(1)计算基组选择不同对体系总能的量子化学计算精度不同, 并且无论选取何种基组进行计算, 量子化学的计算法都存在近似误差;
(2)结构优化的迭代收敛标准设定不同, 优化后结构的无扰体系总能不同。 越严格的收敛标准能够得到越低的体系总能量, 但迭代次数和需要的数据存储空间越大。
4 结 论
以实验法获取了苏氨酸的太赫兹吸收谱, 构建了不同的苏氨酸分子构型, 以量子化学方法模拟计算不同分子构型的太赫兹吸收谱, 并对计算谱进行了讨论。
研究表明, 苏氨酸单分子构型的计算谱与实验谱差异较大, 但单分子构型在高频段吸收峰峰位基本吻合。 二聚体构型的计算谱虽然吸收峰个数与实验谱一致, 但吸收峰位出现红移, 说明考虑了分子间氢键的二聚体构型仍与实际样品结构不符。 晶胞构型的计算谱无论是吸收峰个数还是吸收峰峰位均与实验谱较吻合, 表明样品的太赫兹吸收谱是分子内原子及原子团作用、 分子间氢键及范德华力作用的集中体现。
基于太赫兹时域光谱技术的氨基酸定性检测需要像“查字典”一样依赖于标准吸收谱, 标准吸收谱收录于标准图库, 这些吸收谱都是由实验法获得的, 但实验谱的准确性依赖于实验仪器的精度、 数据收集及处理的舍入误差等, 基于量子化学模拟计算得到的计算谱可以为实验谱提供理论支持。 而某些化学分析需要从微观入手, 这需要找到保持待分析物质理化性质的最小结构, 用实验谱指认计算谱, 可以得到保持苏氨酸最小物理性质的构型为晶胞。
猜你喜欢
动物营养学报(2022年12期)2023-01-05
高中数理化(2022年16期)2022-09-14
高中数理化(2022年4期)2022-03-14
中学生数理化(高中版.高考理化)(2019年10期)2019-11-08
山东化工(2019年1期)2019-01-24
雷达学报(2018年1期)2018-04-04
雷达学报(2018年1期)2018-04-04
雷达学报(2018年1期)2018-04-04
广东饲料(2016年1期)2016-12-01
湖南饲料(2015年1期)2015-04-07
