
氧化三甲胺通过促进成年小鼠心肌细胞T小管重构加重心力衰竭
2020-07-06 03:55纪方方左安俊刘荟婷王庆尧
中国病理生理杂志 2020年6期
靳 步,纪方方,左安俊,刘荟婷,祁 琳,何 韵,王庆尧,赵 鹏△
(1青岛大学附属医院病理科,山东青岛266000;2青岛大学医学部基础医学院,山东青岛266071;3山东省青岛疗养院,山东青岛266071;4青岛大学附属医院普外科,山东青岛266000)
心力衰竭(heart failure,HF;简称心衰)是导致心血管疾病患者死亡的主要原因,绝大部分的心血管疾病最终都会导致心衰[1-2],因此心衰病理机制的研究对发现新的临床诊疗方法具有重要意义。
近年来的研究发现,肠道微生物群参与了许多疾病的发病过程,如肥胖、糖尿病、胃肠疾病、癌症、心血管疾病(包括心衰)等[3-4]。目前的心衰肠道假说认为,心衰可导致细菌移位,增加全身循环中细菌的产生,增加炎症状态,从而导致心衰的进一步发展。氧化三甲胺(trimethylamineN-oxide,TMAO)是由肠道微生物群的代谢产物,肠道微生物群组成的改变会导致TMAO水平升高,产生循环内毒素,进而发展为心衰[5-7]。动物实验表明,在心衰小鼠的心肌细胞中,微管蛋白(tubulin)上调、T小管重构是一个重要和常见的现象[8]。微管是由α-和β-微管蛋白二聚体聚合而成的普遍存在的细胞骨架纤维,而TMAO能够促进微管蛋白聚合[9],微管聚合可导致连接蛋白家族成员的亲联蛋白2(junctophilin-2,JPH2)再分布,从而导致心脏T小管与肌质网的错位,造成心肌收缩功能减退[10]。在一项有1 020名受试者的研究中,通过心脏导管检查发现在进行了冠状动脉疾病血管造影的患者中,TMAO循环水平显著升高[11]。此外,在另外一组正在进行心导管检查的受试者中(n=4 007),TMAO水平升高独立地预测了不良心血管结局的风险,如心脏病发作、中风和死亡风险[7]。在心衰患者中,TMAO血浆水平升高(n=720),升高的TMAO水平与5年死亡风险增加有关[4]。
目前,肠道微生物群失调在心衰发生发展中的作用有待进一步研究,我们推测TMAO的升高可能导致T小管重构和Ca2+处理紊乱,从而损害心脏收缩功能。本研究探讨TMAO对成年小鼠心室肌细胞微管蛋白和T小管结构的影响,为肠道菌群紊乱在心功能下降及心力衰竭中的作用机制提供参考资料。
材料和方法
1 成年小鼠心室肌细胞分离培养
SPF级C57BL/6小鼠,雄性,2~3月龄,体重25~35 g,购自北京维通利华实验动物技术有限公司,许可证号为SCXK(鲁)2019-0003。小鼠腹腔内注射戊巴比妥钠(100 mg/kg),使用Langendorff法心脏灌流[0.5 g/L的2型胶原酶(Worthington)和0.08 g/L的XVI型蛋白酶(Sigma-Aldrich)]12 min到15 min;实验采用≥75%Ca2+耐受杆状肌细胞。在含1.0 mmol/L Ca2+的台氏液[其组成(mmol/L):137 NaCl,5.4 KCl,10葡萄糖,10 HEPES,0.33 NaH2PO4,1 Mg-Cl2;用氢氧化钠将pH值调整至7.35~7.45]中处理10 min,细胞在含有基本培养液(MEM)与10%胎牛血清的经层粘连蛋白(10 mg/L)预处理的T25瓶中培养[12]。将细胞置于37℃、5%CO2培养箱中培养 2 h,后将培养液改为不含血清的MEM,培养24 h。
2 细胞处理
细胞经不含血清的MEM培养24 h后,再将细胞分别置于含有0.3、1、3和 10 μmol/L TMAO(Sigma-Aldrich)的条件下培养24 h,分析TMAO对心肌细胞T小管的影响,并选择一个合适的浓度进行后续实验。
3 方法
3.1 T小管成像与分析 采用Di-8-ANEPPS(10 μmol/L;AAT BioQuest)在无钙台氏液中室温染色30 min。用LSM 510型共聚焦显微镜(Carl Zeiss MicroImaging Inc.)的63倍透镜观察T小管结构。采用IDL图像分析程序对T小管完整性进行定量分析,使用AutoTT软件分析T小管功率(T-tubule power,TTpower)的强弱来反映T小管组织的规整性[9]。
3.2 Western blot 心肌细胞用冷PBS冲洗3次,悬浮于裂解液[其组成(mmol/L):50 Tris-HCl(pH 7.4),150 NaCl,10 NaF,1钒酸钠,5 EGTA,5 EDTA;还有0.2%Triton X-100,0.5%脱氧胆酸钠,0.1%SDS]中。将裂解液13 000×g、4℃离心15 min,上清液作为全细胞蛋白保存。采用BCA法(Pierce)测定蛋白浓度。全细胞蛋白样品在10%的双三聚体凝胶上电泳。电泳后将蛋白转移至PVDF膜(Millipore),并在 4℃条件下使用 JPH2(1∶1 000;Santa Cruz)、calpain I(1∶5 000;Cell Signaling Technology)和 GAPDH(1∶5 000;Sigma-Aldrich)的 I抗过夜。PBS洗涤3次后,用辣根过氧化物酶偶联的II抗(在PBS溶液中1∶50 000~1∶10 000稀释)继续孵育。免疫反应采用增强化学发光检测试剂盒(Pierce)和LAS-3000科学成像系统(FujiFilm)进行可视化。使用NIH ImageJ 1.43d软件对条带进行量化。
3.3 游离和聚合微管蛋白的测定 使用微管/微管蛋白体内检测试剂盒(Cytoskeleton Inc.)检测游离和聚合的微管蛋白。在细胞裂解和微管稳定缓冲液(含 5 mmol/L MgCl2、1 mmol/L EGTA、0.1 mmol/L GTP、1 mmol/L ATP、100 mmol/L PIPES、30% 甘油、0.1%壬酸酯-p40、0.1%Triton X-100、0.1%吐温-20、0.1%β-巯基乙醇、0.001%抗泡剂和0.2%蛋白酶抑制剂,pH 7.4)中均质化细胞。匀浆通过离心机(37℃、100 000×g离心30 min)产生浮层包含自有的微管蛋白和聚合颗粒微观沉淀,用200 μmol/L氯化钙对聚合微管颗粒进行重悬。
3.4 心肌细胞免疫荧光染色 分离的心肌细胞在4%多聚甲醛中37℃固定15 min,用PBS冲洗3次,每次10 min,然后在含有0.3%Triton-X 100的PBS中渗透30 min。在室温下1%BSA封闭30 min后,β-tubulin抗体(1∶100;Sigma)和JPH2抗体(1∶50;Santa Cruz)4℃条件下孵育过夜。I抗孵育过后,与荧光标记的II抗在室温下孵育2 h。用63倍透镜共聚焦显微镜观察。用ImageJ 1.43d软件定量微管密度,用IDL图像分析程序分析JPH2结构(JPH2 power)。
3.5 共聚焦Ca2+成像 将心肌细胞置于含有5 μmol/L Fluo-4 AM(Invitrogen)和1.8 mmol/L Ca2+的台氏液中预处理30 min,然后用无染料的台氏液洗涤细胞20 min,使其脱酯化,然后成像处理。采用共聚焦显微镜沿肌细胞纵轴进行扫描,获取Ca2+图像。1 Hz磁场刺激下,在含有1.8 mmol/L Ca2+的台氏液中测量稳态Ca2+瞬态,停止磁场刺激后5 s左右记录Ca2+火花。8个像素中每个扫描像素的触发时间的平均绝对偏差为Ca2+瞬变失同步化指数,用来评估Ca2+瞬态的失同步性[13]。
3.6 小鼠心功能检测 将10只雄性SPF级C57BL/6小鼠分为TMAO组与对照组,每组各5只。将0.12%TMAO添加到TMAO组小鼠饲料中,6周后与喂养正常鼠粮的对照组小鼠一同进行心功能检测。
4 统计学处理
数据用均数±标准差(mean±SD)表示。多组比较采用单因素方差分析;两组比较采用t检验。采用SPSS 15.0软件进行统计分析。以P<0.05为差异有统计学意义。
结 果
1 TMAO导致心肌细胞T小管网络损伤
在检测浓度(0.3~10 μmol/L)范围内,TMAO对心肌细胞T小管组织的损伤呈浓度依赖性,3 μmol/L和10 μmol/L的TMAO更显著地降低了T小管密度和TTpower(P<0.01),见图1。基于以上结果,我们选择3 μmol/L的TMAO用于后续实验。
2 TMAO导致钙离子处理功能障碍
心肌细胞经TMAO(3 μmol/L)培养24 h后,Ca2+瞬变平均振幅显著降低,到达Ca2+瞬变峰值的时间显著延长,失同步化指数显著增大(P<0.01),见图2。此外,TMAO处理导致自发Ca2+火花频率和Ca2+火花振幅显著增加(P<0.01),但Ca2+火花平均时间半高宽和Ca2+火花半峰全宽无明显改变,见图3。
3 TMAO诱导心肌细胞JPH2从T小管向外周质膜再分布
与对照组相比,TMAO作用24 h后心肌细胞的JPH2分布发生改变。主导频率下JPH2功率的峰值是反映JPH2规律性的定量指标。计算结果显示,TMAO组JPH2功率降低(P<0.01),见图4A、B。此外,经TMAO处理的心肌细胞在心肌细胞边缘可见明显的JPH2堆积(P<0.01),见图4A、C。与对照组相比,TMAO组心肌细胞JPH2蛋白表达水平无明显变化(P>0.05),见图4D、E。
4 TMAO促进心肌细胞微管的聚合
与对照组相比,经TMAO处理后的心肌细胞微管网络密度显著升高(P<0.01),见图5A。Western blot结果显示,经TMAO处理后的小鼠心肌细胞微管聚集性显著增强(P<0.01),见图5B。
5 TAMO损害小鼠心功能
超声心动图显示,TMAO组小鼠心脏收缩功能(左心室射血分数)较正常喂养的对照组小鼠显著下降(P<0.05),见图6。
讨 论
心血管疾病(cardiovascular diseases,CVD),包括动脉粥样硬化、高血压、心力衰竭、房颤、心肌纤维化等,与世界范围内的高死亡率相关。吸烟、不良的饮食习惯、肥胖、糖尿病、高胆固醇等是众所周知的危险因素[1]。近年来的研究表明,肠道微生物群及其代谢产物在CVD的发生发展中起着关键作用[14]。动物模型证实了胆碱、肠道微生物群和TMAO加速了动脉粥样硬化、心力衰竭等CVD的发展,并与疾病严重程度有关。在动物实验中,经TMAO灌胃后的小鼠,心脏体积明显增大[15]。临床数据也表明,心力衰竭患者血浆中肠道微生物源代谢物TMAO浓度呈现升高趋势[4]。心肌兴奋-收缩偶联功能的关键是T小管和肌质网之间的空间关系,在心脏动作电位过程中,Ca2+通过L型Ca2+通道激活邻近肌质网膜上的雷诺丁受体(ryanodine receptors,RyRs),从肌质网释放更多的Ca2+[16]。T小管存在于正常成年哺乳动物的心肌细胞中,与细胞外空间连续,并深入到哺乳动物心室肌细胞的内部[17]。在各种心脏疾病,包括不同类型的心力衰竭和心肌病等情况下,T小管会发生显著的重塑或丢失。

Figure 1.Effects of TMAO on T-tubule structure in adult mouse cardiomyocytes(Di-8-ANNEPS staining,×63).Isolated adult mouse cardiomyocytes were exposed to increasing concentrations of TMAO for 24 h.The global T-tubule density and T-tubule power(TTpower)were determined.Mean±SD.There were 25~30 cells in each group.*P<0.05 vs control group.图1 TMAO对成年小鼠心肌细胞T小管结构的影响
TMAO和心衰之间的机制是否与心肌细胞T小管重构有关,目前尚不清楚。因此,在我们的研究中,我们将分离的成年小鼠心肌细胞利用TMAO处理,来确定TMAO是否会破坏T小管结构以及T小管丢失或(和)紊乱的程度。结果显示,暴露于TMAO条件下24 h的小鼠心肌细胞T小管结构紊乱,经TMAO处理后的小鼠心肌细胞T小管密度及TTpower显著下降。由于Ca2+从肌质网释放是由Ca2+通过细胞膜内流触发的,而T小管重构破坏了位于T小管膜上的电压门控L型Ca2+通道与肌质网上的Ca2+释放通道(RyR2)之间的精确通信,从而干扰肌质网Ca2+释放的快速电激发、启动和同步触发[18-19]。因此,我们研究了TMAO是否能诱导分离的心肌细胞Ca2+处理紊乱。结果显示,经TMAO培养的小鼠心肌细胞到达Ca2+瞬变峰值的时间显著延长,Ca2+瞬变同步化减少,自发Ca2+火花频率和Ca2+火花振幅也明显增加。这些数据表明,TMAO能够破坏心肌细胞Ca2+处理和干扰Ca2+再利用。Ca2+释放的同步化保证了心室内的细胞收缩是协调一致的,随着心室压力的增加和体积的减小,心肌的收缩会把血液挤出心脏。哺乳动物的心室肌细胞几乎同步从肌质网触发释放Ca2+,引起心肌细胞去极化,而主要负责每个细胞内局部Ca2+释放同步化的就是T小管系统。Ca2+非同步化释放常以Ca2+触发释放的传播延迟为特征,心肌细胞到达Ca2+瞬变峰值的时间延长,导致细胞收缩和(或)Ca2+瞬变功能障碍。
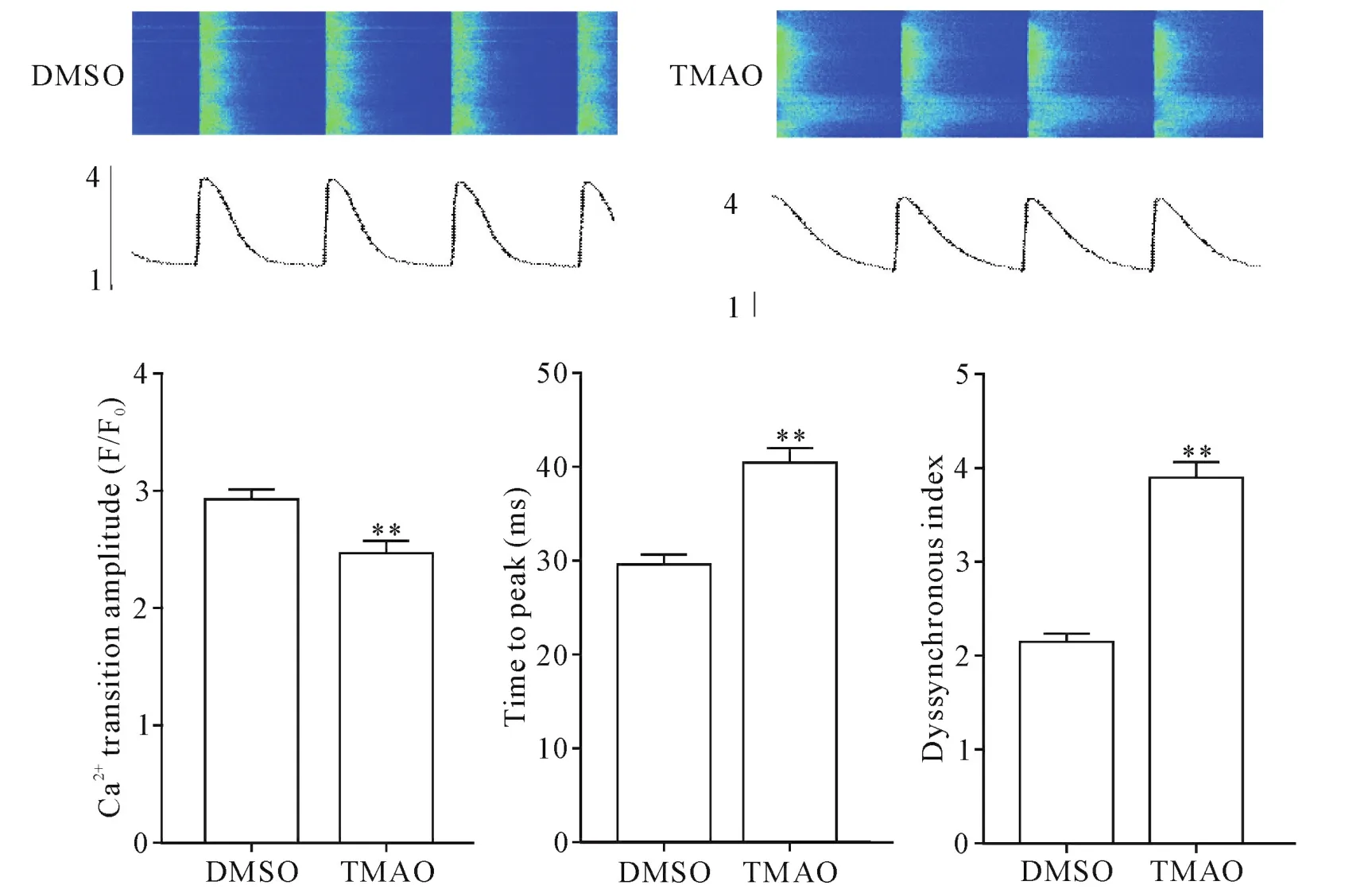
Figure 2.TMAO impaired Ca2+handling in the cardiomyocytes.Mean±SD.There were 25~30 cells in each group.**P<0.01 vs control group.图2 TMAO抑制心肌细胞Ca2+处理

Figure 3.Cardiomyocytes treated with TMAO displayed more Ca2+sparks(Fluo-4 AM staining,×63).FDHM:full duration at half maximum;FWHM:full width at half maximum.Mean±SD.There were 15~30 cells in each group.**P<0.01 vs DMSO group.图3 经TMAO处理的心肌细胞产生更多的Ca2+火花
T小管重构是在不同病因的晚期心力衰竭中一个重要和常见的现象,T小管重构导致的Ca2+释放不同步使心肌收缩力降低[8]。随着T小管完整性的丧失,收缩期Ca2+瞬时变化的“钙触发钙释放”模式发生干扰。T小管断裂,L型Ca2+通道与RyR2发生错位,导致“孤立的”RyR簇,从而增加了Ca2+火花诱导Ca2+火花发生的机会[20-21]。Na+-Ca2+交换器的缺失以及因此而导致的更广泛的Ca2+细胞分布可能使大型哺乳动物的心室或心房易于发生Ca2+依赖的延迟去极化,这可能是大型哺乳动物心室肌细胞Ca2+超载机制的基础[22-23]。局部的Ca2+超载可能导致异常Ca2+释放,这可能是导致心肌收缩功能障碍的原因。
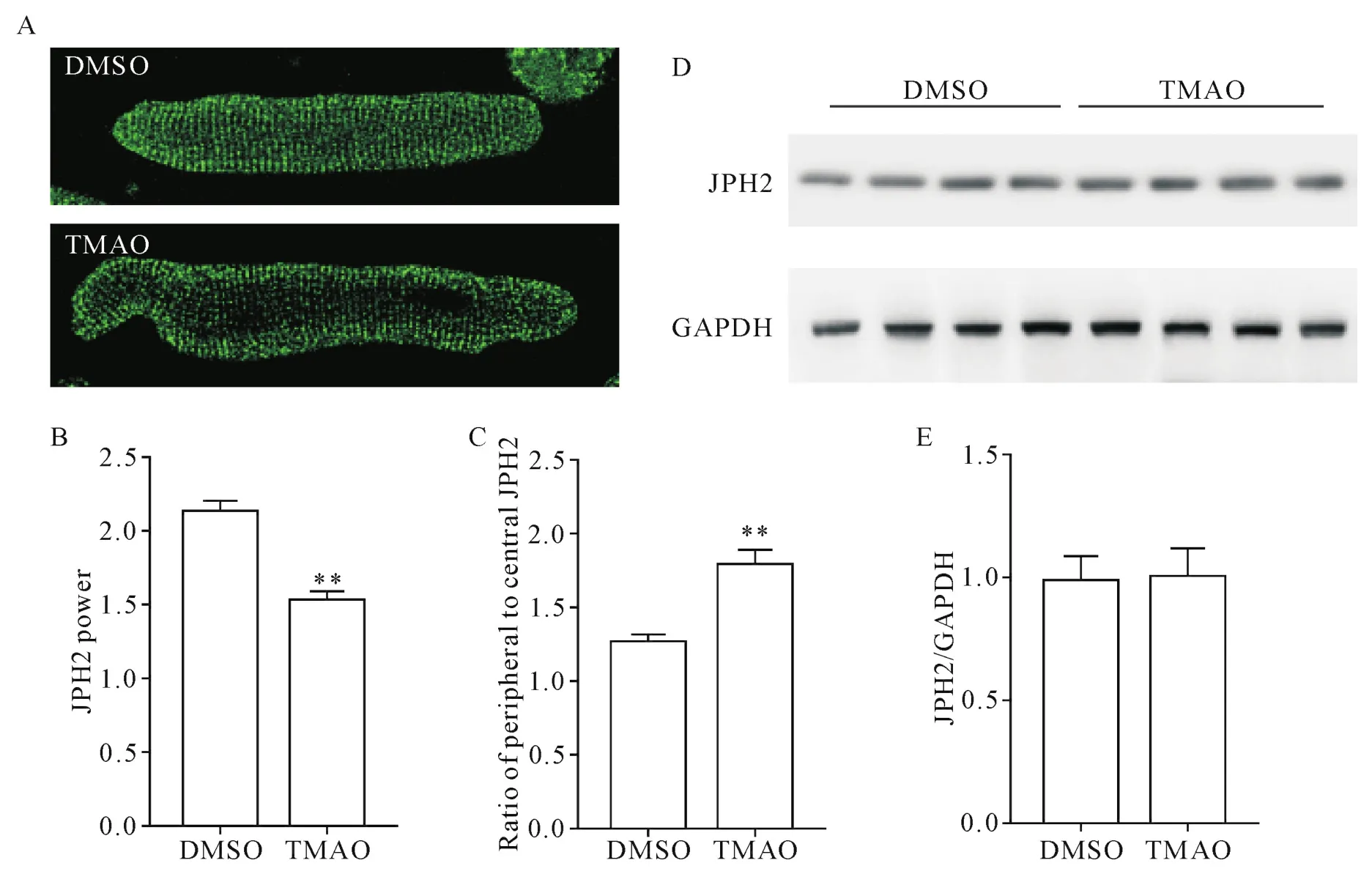
Figure 4.TMAO aggravated JPH2 translocation in the cardiomyocytes.A:representative images of isolated cardiomyocytes stained with anti-JPH2 antibody after treatment with or without TMAO for 24 h(×63);B:JPH2 power;C:semi-quantitative analysis of JPH2 distribution between T-tubule and peripheral sarcolemma;D:representative image of Western blot;E:densitometric data of Western blot.Mean±SD.There were 15~30 cells in each group.**P<0.01 vs DMSO group.图4 TMAO加重了心肌细胞中JPH2的易位

Figure 5.More rearrangement and densification of microtubules in the cardiomyocytes with TMAO treatment.A:β-tubulin immunofluorescence staining in cells treated with or without TMAO(×63);B :Western blot of free(F)and polymerized(P)β-tubulin.Mean±SD.There were 20~25 cells in each group.**P<0.01 vs DMSO group.图5 在TMAO处理下,心肌细胞的微管发生更多重排和致密化
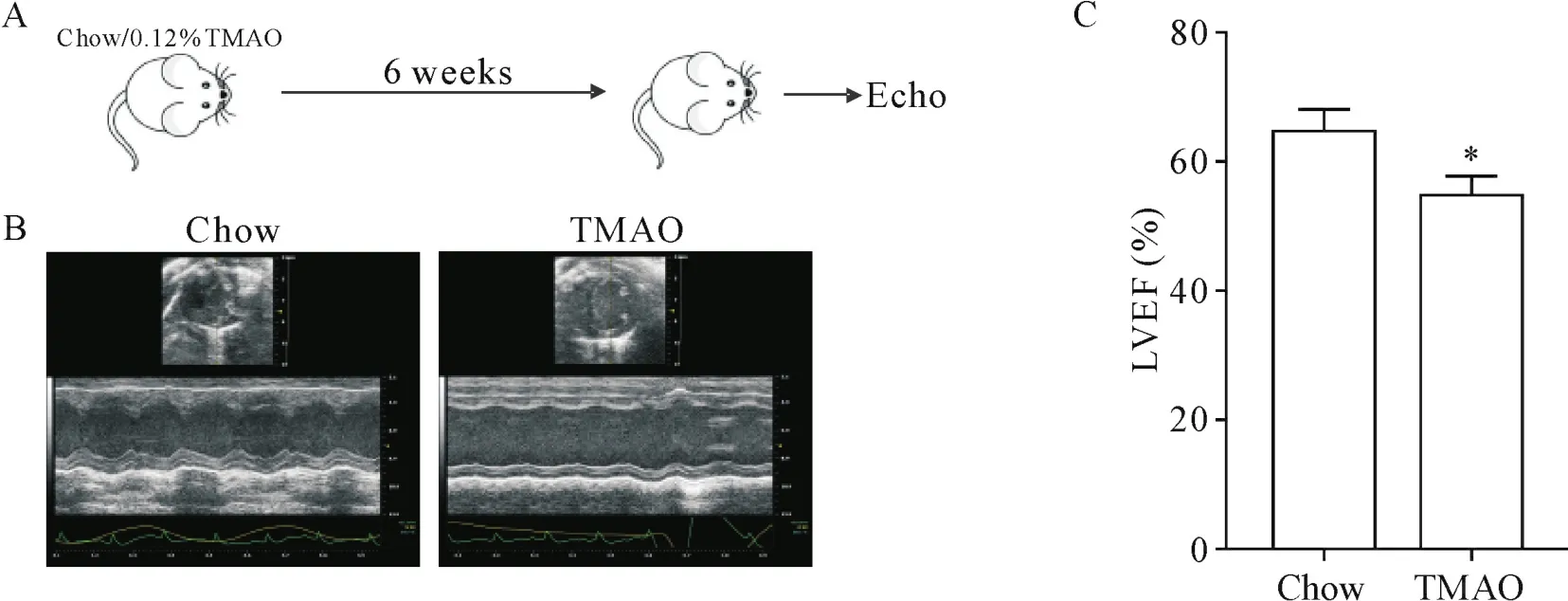
Figure 6.TMAO undermined mouse cardiac function.A:the protocol showed how to treat mice with TMAO and assay the left ventricular ejection fraction(LVEF);B:representative short-axis echocardiographic images of left ventricle M-mode recordings from chow-and TMAO-treated mice;C:quantitative evaluation of LVEF.Mean±SD.n=4~5.*P<0.05 vs chow group.图6 TMAO破坏小鼠心脏功能
维持T小管组织的关键是JPH2,它将T小管连接在肌质网上,保障心脏二联体的结构[24]。JPH2在兴奋细胞中连接细胞膜和肌质网的物理间隙,维持正常心室肌细胞的连接膜复合体[25]。JPH2基因敲除对胚胎是致命的;而敲减JPH2基因的胚胎,其肌细胞存在缺陷的心脏二联体,包括更多没有JPH2的肌质网片段[26]。研究还表明,JPH2在心力衰竭动物模型中呈现重新分布和减少[16]。因此,我们推测JPH2易位和(或)下调可能在TMAO处理下引起的T小管重构中发挥重要作用。我们的结果表明,TMAO作用小鼠心肌细胞24 h后,JPH2分布发生改变,在心肌细胞边缘可见明显的JPH2堆积,提示TMAO处理后JPH2蛋白分布发生易位;此外,TMAO组的JPH2功率也降低。以上结果提示了JPH2蛋白易位可能是TMAO诱导的T小管紊乱的主要潜在机制。
由于微管表达的增加和稳定微管的形成,微管在代偿期和失代偿期积累并致密化。通过JPH2的再分布,微管致密化可导致心脏疾病和心肌细胞T小管重构。在一些研究中,TAMO的增加已成为预测心力衰竭严重程度的重要指标之一。数据表明,TMAO确实促进了微管蛋白聚合,并稳定了微管,使微管不受失稳扰动[9]。因此,我们研究了TMAO是否参与导致微管积聚和兴奋-收缩偶联功能障碍,结果显示,小鼠心肌细胞经TMAO处理后,与对照组相比,微管网络的致密化和聚集性显著增强。先前的研究表明,微管蛋白致密化介导了JPH2在心脏应激反应中的易位现象[16],这与我们的研究结果一致,即由于微管蛋白聚合增加,JPH2在TMAO处理中发生错位。JPH2在维持成年心脏心肌细胞T小管结构完整性和维持 Ca2+处理方面是必需的[10,21]。我们的实验结果显示,TMAO诱导的JPH2易位促进了T小管重塑,并且动物实验的结果也表明TMAO能够使小鼠心功能下降。
总之,本研究为TMAO如何通过促进小管蛋白的形成和随后JPH2的易位而损害心功能提供了证据。这些实验结果表明,减少TMAO的产生,如改善肠道菌群的质量或防止肠道菌群失调,可能在一定程度上能够保护心功能和延缓心衰的发展。
猜你喜欢
中国骨质疏松杂志(2022年9期)2022-10-18
——水芹主要害虫识别与为害症状
长江蔬菜(2022年13期)2022-07-29
土壤与作物(2021年2期)2021-06-01
青岛大学学报(医学版)(2020年6期)2020-11-16
青岛大学学报(医学版)(2020年3期)2020-07-04
青岛大学学报(医学版)(2020年3期)2020-07-04
辅导员(2020年6期)2020-04-23
中国癌症防治杂志(2019年5期)2019-01-04
派出所工作(2018年4期)2018-09-10
江苏农业科学(2016年12期)2017-04-05
