
IgG4相关性疾病1例
2020-07-05 05:59:22周云姜泓黄长形
肝脏 2020年6期
周云 姜泓 黄长形
患者,女性,65岁,主 “口苦、尿黄1月余”于2019年6月入院。患者4个月前因“高血压”在当地中医诊所口服中药治疗4个月。1个月前自觉口苦、尿黄,伴右上腹胀、乏力。无发热、腹痛,无恶心、呕吐。就诊于我院门诊,查肝功ALT 641 U/L,AST 723 U/L,ALB 33.8 g/L,T-Bil 85.4 μmol/L,D-Bil 34.5 μmol/L,腹部B超示肝脏大小正常,肝囊肿,胆囊大小正常,胆固醇结晶析出,脾略大,脾内钙化灶,胰腺大小正常。胃镜提示:慢性非萎缩性胃炎,十二指肠球部溃疡。门诊以“黄疸待查”收住院。查体:全身皮肤巩膜中度黄染,肝掌阴性,未见蜘蛛痣。心肺查体无明显异常。腹部平坦、柔软,无腹壁静脉曲张。右上腹部压痛,无反跳痛。肝脾肋下未触及,肝区叩痛阴性,移动性浊音阴性,肠鸣音4次/min,双下肢轻度水肿。入院后查血常规、凝血功能、肿瘤标志物无异常。肝功 ALT 511 U/L,AST 493 U/L,ALB 31.2 g/L,T-Bil 100.9 μmol/L,D-Bil 63.3 μmol/L,免疫球蛋白补体系列IgG 26.6 g/L,IgA 4.45 g/L,κ-LC 6.82 g/L,λ-LC 3.16 g/L,C3c 0.76 g/L,IgG亚型IgG1 20.6 g/L(参考值4.05-10.11 g/L),IgG2 4.38 g/L,IgG3 0.31 g/L,IgG4 >3.6 g/L(参考值0.03~2.01 g/L),抗核抗体1:100弱阳性,乙肝五项阴性,抗HCV阴性,甲肝抗体、戊肝抗体阴性,自身免疫性肝抗原谱阴性,系统性多血管炎相关抗体测定阴性。心肌酶谱 CK 757 U/L,CK-MB 37 U/L,LDH 422 U/L,α-HBDH 280 U/L。影像学检查:腹部CT示肝硬化,肝左叶外侧段及肝右叶囊肿,肝右叶后下段钙化灶,肝实质密度欠均匀,建议行增强检查,胆囊炎,脾大,双肾低密度影,考虑囊肿(图1)。腹部MR及MRCP示肝实质弥漫性异常改变,呈明显强化,结合临床及病史,结缔组织病变不除外(图2)。
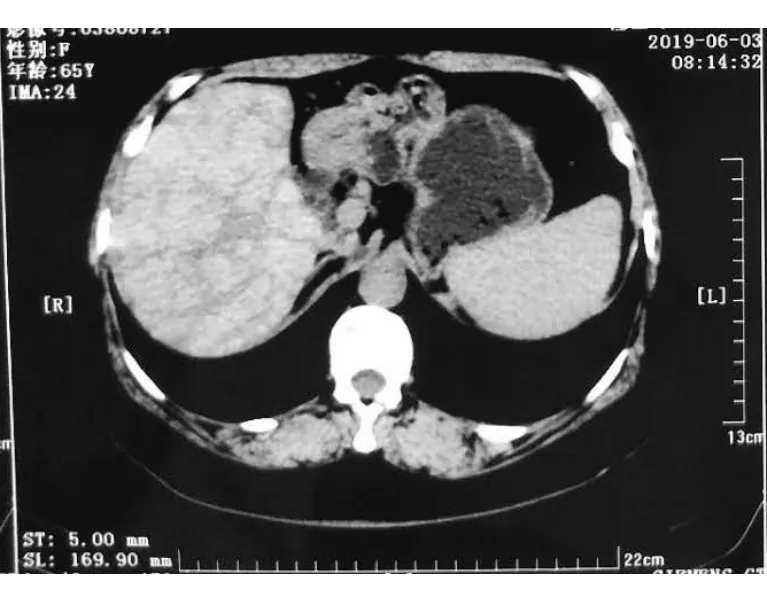
图1 腹部CT图
肝内胆管、左右肝管、肝总管及胆总管未见明显扩张,胆囊腔内未见异常信号,胰腺大小、形态尚可,信号未见异常,胰管未见扩张(图3)。
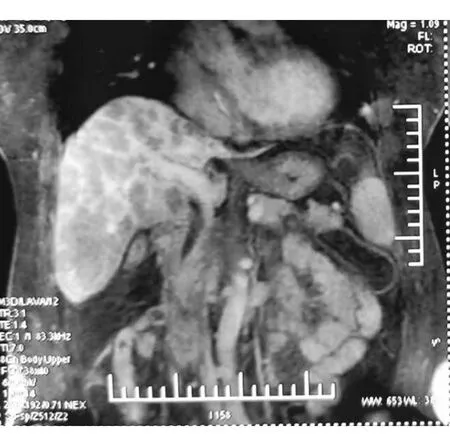
图2 腹部MR图
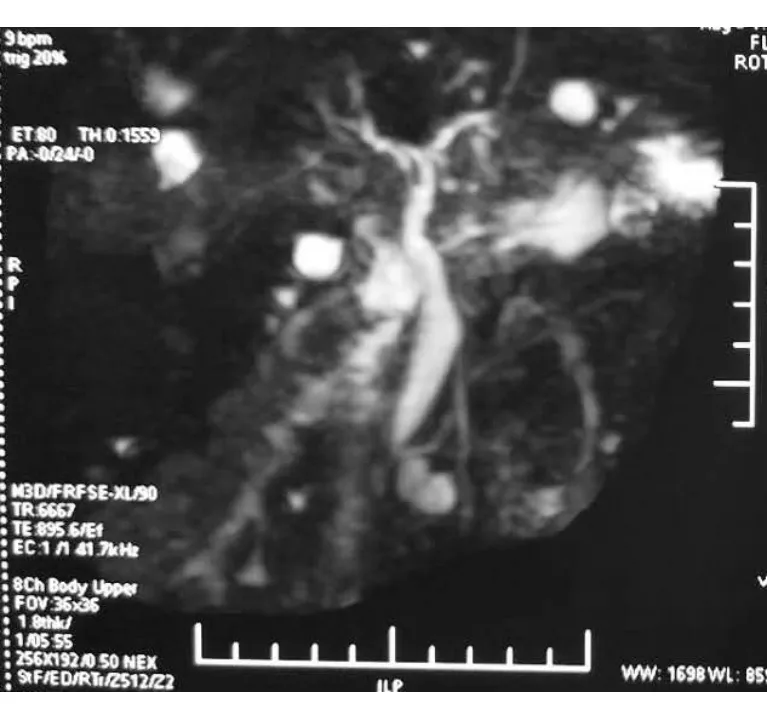
图3 MRCP图
入院后给予丁二磺腺苷蛋氨酸、甘草酸单铵半胱氨酸保肝退黄治疗,并给与地塞米松钠 10 mg 3 d,5 mg 2 d抗炎治疗。诊断IgG4相关性疾病后给与泼尼松 40 mg/d治疗。复查肝功 ALT 183 U/L,AST 51 U/L,Alb 30 g/L,T-Bil 46.5 μmol/L,D-Bil 29.6 μmol/L,病情好转出院,嘱其口服泼尼松40 mg/d治疗,逐渐减量,并5 mg/d维持6个月。
讨论IgG4相关性疾病(IgG4-related disease,IgG4-RD)是一种慢性炎症性自身免疫性疾病,常见于中老年男性,主要表现为受累脏器的肿胀或者炎性假瘤形成,以血清IgG4水平增高,受累组织中富含主要分泌IgG4的淋巴浆细胞为特征[1]。2011年日本风湿病学会IgG4-RD诊断标准为[2]:(1)在包括泪腺、唾液腺、胰腺、淋巴结、肺在内的多器官系统的单发或多发肿块、硬化或肿块,(2)血清IgG4水平≥135 mg/dL,(3)组织病理学发现高的淋巴细胞、浆细胞浸润、明显纤维化(IgG4+/IgG+ >40% 且>10个IgG4阳性的浆细胞/HPF)。IgG4-RD通常被认为是一种罕见的疾病,但其真正的流行病学尚未完全阐明,近年来该病的发生率增高。IgG4-RD的诊断工作非常复杂,通常需要结合临床检查、影像学、组织学和血清学分析。其诊断需要结合患者的临床表现仔细解读检查结果,排除各种鉴别诊断。在过去的几年里,这一疾病新的生物标志物和辅助检查,包括IgG4+浆细胞计数、IgG4反应指数(IgG4-responder index ,IgG4 RI),CT、MRI、PET/CT检查和新的治疗方法(利妥昔单抗)被提出,拓宽了IgG4-RD领域的知识[3]。据一个日本的研究报道[4],70%的IgG4-RD患者因出现症状被诊断,而30%是偶然发现的。最常见的症状与肿胀有关,如梗阻性黄疸和眼球突出(41%),其次是一般腹部症状(18%)和不适(4%)。肾脏(4%)和肺部症状(3%)只出现在少数患者中。lgG4-RD诊断前另一个经常报告的症状是体重减轻。
IgG4-RD累及肝脏的疾病被命名为IgG4相关肝病,但相关报道非常少见。IgG4相关肝病患者的肝实质内可见平均2~60/HP的IgG4阳性浆细胞广泛浸润。 Umemura等[5]根据患者肝脏组织病理学特征将IgG4相关肝病分为以下五种类型:(1)门静脉炎症模式:门静脉炎症明显,有或无界面性炎症;(2)大胆管损伤模式:胆管增生、中性粒细胞浸润和门静脉区域水肿;(3)门静脉硬化模式:表现为密集的门静脉硬化、门静脉炎症瘢痕;(4)小叶型肝炎模式:小叶内水肿,肝细胞坏死,类似病毒性肝炎;(5)胆汁淤积型模式:主要为肝小叶中心区的小管型胆汁淤积。
该病用糖皮质激素(glucocorticoids,GC)治疗有效,泼尼松30~40 mg/d或0.6 mg/(kg·d),根据病情逐渐减量至2.5~5 mg/d维持,约98%的患者可缓解。24%~33%的患者药物减量后有疾病复发,可加用硫唑嘌呤或环磷酰胺等免疫抑制剂[3]。某前瞻性群组研究纳入122例新诊断为IgG4-RD的患者,进行糖皮质激素(GC)单药或GC联合免疫抑制剂治疗,随访至少3年。研究发现在激素减量和维持阶段,联合应用免疫抑制剂比单纯应用GC更有效。血清IgG4水平升高、累及更多器官、IgG4 RI评分升高、过敏史、基线时嗜酸性粒细胞升高、血清IgG4水平重新升高、随访时GC维持剂量降低可能预示复发[6]。
总之,IgG4-RD最常见于中老年男性,具有受累器官亚急性包块生成或器官弥漫性肿大、淋巴结肿大、血清IgG4水平升高以及特定器官病变等临床特征。诊断依据是特征性组织病理学活检结果,单纯血清学指标不具有诊断作用,但具有重要辅助作用。糖皮质激素起始治疗效果良好也是IgG4-RD 的特征之一,停止治疗后常见复发,需进一步关注患者的恶性肿瘤风险[7]。本例患者有服中药史,排除病毒性肝炎后首先考虑药物性肝损害,然影像学提示已有肝硬化,与病程不符,进一步查IgG4得以明确诊断。临床上不明原因的肝功能异常,应完善IgG4检查,避免漏诊,延误治疗。
猜你喜欢
中国药学药品知识仓库(2022年2期)2022-03-23 00:19:16
中国临床医学影像杂志(2021年5期)2021-08-13 09:01:38
天津医科大学学报(2021年3期)2021-07-21 09:04:00
中国临床医学影像杂志(2019年6期)2019-08-27 02:59:46
中国生物医学工程学报(2019年5期)2019-07-16 07:56:42
中国医学影像学杂志(2018年9期)2018-10-17 01:27:04
中国中西医结合皮肤性病学杂志(2016年4期)2016-07-18 10:59:52
实用皮肤病学杂志(2015年4期)2015-12-22 11:21:40
西南军医(2015年3期)2015-04-23 07:28:36
卫生职业教育(2014年8期)2014-02-16 08:00:38
