
泛酸激酶相关神经变性病2 例报告并文献复习
2020-06-30 03:17:52杜祥慧
临床儿科杂志 2020年6期
杜祥慧 郭 虎 何 燕 梁 超
南京医科大学附属儿童医院神经内科(江苏南京 210008)
脑内铁沉积性神经变性病(neurodegeneration with brain iron accumulation,NBIA),又称苍白球黑质红核色素变性,是铁代谢障碍引起的一种罕见的中枢神经系统变性疾病[1]。NBIA 主要表现为锥体外系受累,起始为姿势异常、肌张力增高、步行困难和不自主运动,症状呈进行性加重,无特效疗法。近年来发现,大多数NBIA患者都存在泛酸激酶2(PANK2)基因突变[1],存在或被怀疑存在PANK2基因变异的NBIA被称为泛酸激酶相关神经变性病(pantothenate kinase associated neurodegeneration,PKAN)[2]。本文报告2例PANK2基因变异的NBIA患儿的临床资料及基因分析结果,并进行相关文献复习,为临床诊断和遗传咨询提供依据。
1 临床资料
例1,男性,7岁2月龄,因肌张力升高3年余、加重2周于2018年7月就诊于南京医科大学附属儿童医院神经内科。患儿 G1 P1,足月顺产,否认产伤及窒息史。14月龄可行走5、6步,但站立不稳,易跌倒,智力发育稍落后;2岁5月龄偶有肢体抖动,3岁时有右手背伸,上肢僵硬,精细动作欠佳;5岁10月龄出现浅睡期全身抖动,不能行走,不能独站;6岁1月龄出现双上肢屈曲,双手握拳,双下肢向后伸展,呈强直状,腰背呈反折状,喉口发出声音,持续约10 分钟缓解,每4~5天至半个月1次;7岁1月龄双上肢屈曲、双手握拳、双下肢向后伸展等锥体外系症状加重为每天发作,外力不能改变体位,醒后即见,睡眠状态缓解。体格检查:神清,精神反应一般,卧床,异常姿势体位;语言障碍,问答不能;呼吸平,皮肤、浅表淋巴结无异常;双侧瞳孔等大等圆,对光反射存在,眼球活动不配合;咽稍红,两侧扁桃体无肿大;心肺腹无异常;肌力检查不配合,双下肢肌张力明显增高;睡眠状态下膝反射稍活跃、跟腱挛缩,右侧踝阵挛阳性,双侧巴氏征阳性(图1)。实验室检查:血常规、肝肾功能、心肌酶谱均无异常;脑脊液常规、生化、病理及培养均未见异常,脑脊液病原学检查未见异常。头颅磁共振成像(MRI)检查可见苍白球中央高信号、周围低信号环绕,即虎眼征(图2A~C)。其父、母均未见特殊临床表现,无行走异常、肌张力障碍家族史。
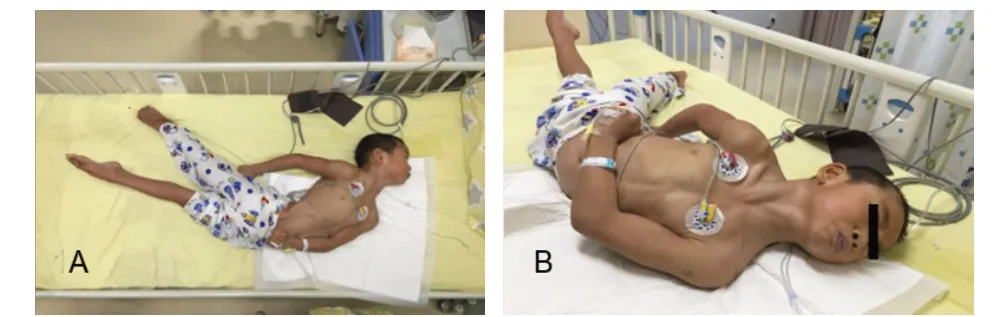
图1 例1 肌张力障碍表现
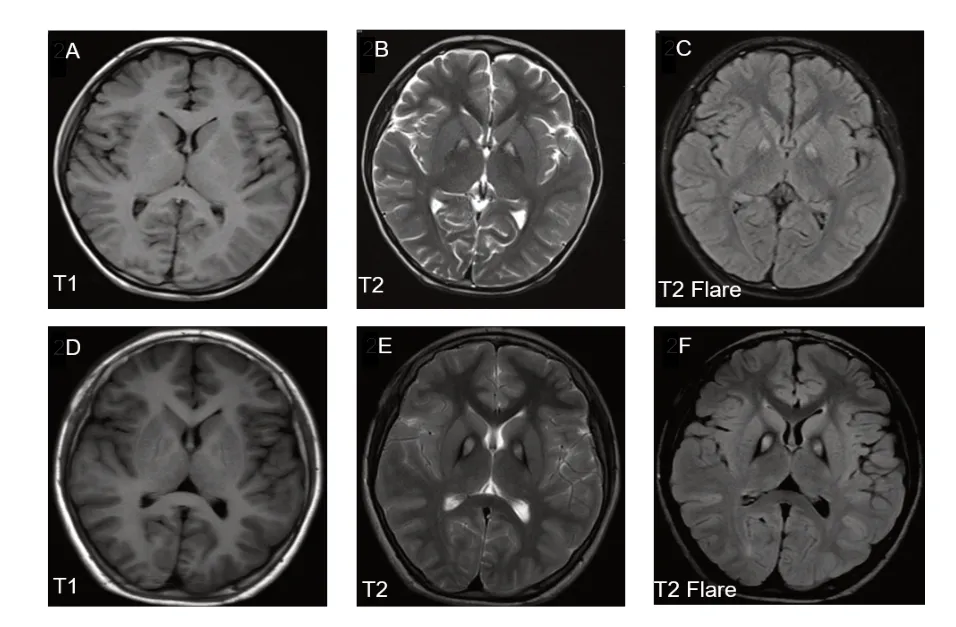
图2 患儿头颅MRI 结果
例2,男性,14岁9月龄,因走路不稳8年、加重1年于2018年3月就诊于南京医科大学附属儿童医院神经内科门诊。患儿G2P2,足月,因羊水早破行剖宫产,否认产伤及窒息史。6~7岁时出现步态不稳,双眼视物不清,学习成绩差,智力欠佳;13 岁时出现口齿不清,走路易摔倒,有不自主头后仰姿势。体格检查:神清,精神反应可,异常走路姿态;构音障碍、言语不清;呼吸平,皮肤、浅表淋巴结无异常;双侧瞳孔等大等圆,对光反射存在,眼球活动正常;心肺腹无异常;肌力检查不配合,四肢肌张力明显增高,膝反射稍活跃,踝阵挛阴性,双侧巴氏征阴性。实验室检查:血常规、肝肾功能、心肌酶谱均无异常。头颅 MRI检查可见“虎眼征”(图2D~F)。其父母未见特殊临床表现;姐姐20岁,身体健康。无行走异常、肌张力障碍家族史。
为明确诊断,经本院医学伦理委员会审核(批准文号:201802001-1),并获得患儿及父母知情同意,采集患儿及其父母外周静脉3 mL,乙二胺四乙酸抗凝,送南京金域医学检验实验室行全外显子基因测序,并根据测序结果,对患儿及父母进行Sanger检测及验证。
例1患儿发现PANK2基因杂合变异C.515-527del、C.644-645delGAinsAT。C.515-527del为杂合致病突变,来源于母亲;C.644-645delGAinsAT为杂合可疑致病突变,来源于父亲(图3A)。C.515_527del系常染色体隐性遗传,该突变为移码突变,预计会使所编码蛋白质自第172位氨基酸,缬氨酸(Val)开始发生移码,并使得蛋白质翻译提前终止,导致所编码蛋白质发生截短从而影响其正常功能。千人基因组和人类基因组突变数据库均未见收录,依据美国医学遗传学与基因组学学会(ACMG)变异分类标准归为“致病性变异”。C.644_645delGAinsAT系常染色体隐性遗传,该突变为错义突变,翻译产物蛋白质第215位氨基酸残基由甘氨酸(Gly)变为天冬氨酸(Asp)。千人基因组和人类基因组突变数据库均未见收录,目前未见文献报道,依据ACMG 变异分类标准归为“可疑致病性变异”。采用 WuXi NextCODE 临床序列分析软件(Clinic Sequence Analyzer,CSA),发现与患儿表型相关的致病基因及具有临床意义的遗传变异。结合父母的检测结果推断两者的变异位于不同等位基因上。例2发现PANK2基因纯合变异,c.397A>G(p.Met133Val)。该变异检测为纯合子,患儿母亲为杂合变异,患儿父亲未检测到该变异(图3B~D ),该基因变异为常染色隐性遗传。千人基因组和人类基因组突变数据库均未见收录。根据ACMG变异分析标准,该位点被定义为临床意义不明偏疑似致病(VUS-LP)变异。
2 讨论

图3 患儿相关基因检测结果
以“Hallervorden-Spalz”、“脑组织铁沉积”、“NBIA”和“PKAN”、“PANK 2”为检索词,分别检索2019 年3月前的PubMed、人类基因组突变数据库、中国知网数据库和万方数据库,检索年限为数据库建库至2019年3月。结果显示,国内外均有关于PANK2基因变异及其临床表现的报道,但基因变异位点相同者较少,本病例报道对于拓展人类基因变异数据库、帮助临床诊断及遗传咨询具有重要作用。
NBIA(曾称为Hallervorden-Spatz综合征)具有较强的临床及遗传学异质性,具体发病机制尚不清楚,但与铁代谢障碍有关,基底节区铁沉积是其最重要的病理特点。根据发病年龄、疾病进展速度及临床表现将NBIA 分为2 型:①经典型,又称为早发型,临床主要表现为锥体外系功能障碍及皮质脊髓束受累的症状与体征,常伴有色素性视网膜病,发病年龄<10岁,病情进展较快,10~15年即失去独立行走能力,20岁前生活不能自理;②不典型,又称为迟发型,发病年龄≥10岁,临床表现除锥体外系功能障碍及皮质脊髓束受累表现外,大多以构音障碍及精神异常为主要首发症状,极少数伴有色素性视网膜病,病情进展缓慢,多数患者到后期仍能行走[3]。本组2例患儿均为早发型,主要临床表现为锥体外系功能障碍,其中例1肌张力障碍表现更为突出并伴有肌张力障碍危象,例2构音障碍、言语不清现象较例1突出。
NBIA 最主要的致病基因是PANK 2基因,约占NBIA的50%[4]。本组2例患儿全外显子测序及父母样本Sanger验证,均检测到PANK2基因变异。PKAN主要临床表现为进行性加重的肌张力障碍、构音不清、手足徐动、强直等锥体外系症状。本组2 例患儿的临床表现与之相符。PANK2基因多为纯合变异,也有少数为复合杂合变异,少数仅发现1 个杂合变异[5]。本组2例患儿中,例1为杂合子,但临床表型重于为纯合子的例2,这可能是由于变异检测条件的限制,某些变异(如启动子区)未被检测到;或者虽然仅有1 个变异的等位基因,但同时在PANK 2催化合成辅酶A(CoA)的下游通路发生功能障碍,由这两种因素相互作用而致病[6-8]。PKAN基因变异类型包括碱基缺失、重复、插入、剪接位点变异和错义变异[9]。从PANK 2基因所表达的蛋白质来讲,一般分为2 种类型:导致氨基酸替代的变异,即错义变异和造成蛋白质截断的无效变异(null mutations),某些变异引起的临床表型轻微[8,10]。
PKAN的病理基础是铁盐不断沉积于苍白球和黑质等部位,导致神经元死亡、胶质细胞增生、水含量增加,以及空泡形成。T2加权成像(T2WI)双侧苍白球呈周围低信号、中央斑点状高信号改变,形似虎眼,即所谓的“虎眼征”。出现“虎眼征”是诊断PKAN 的主要依据之一。本组2 例患儿均出现典型的“虎眼征”。虽然,大多数研究提示典型的“虎眼征”出现于首发症状之后,但是目前关于“虎眼征”在病程中出现的具体时间及规律尚无系统研究。有报道称,“虎眼征”在病程进展期可消失[11]。但以运动障碍,即锥体系及锥体外系损害症状为主的青少年患者,当T2WI扫描呈现“虎眼征”等特异性改变时,即可高度怀疑PKAN。有上述影像学表现,同时兼有阳性家族史者则支持NBIA诊断。反之,对疾病早期头部MRI检查无“虎眼征”的患者,不能排除典型PKAN诊断。随着疾病进展和症状明显,头部MRI可显示出典型的“虎眼征”,但也有些患者的MRI始终无铁沉积表现[12]。PKAN患者颅脑T1加权成像(TIWI)一般无明显异常改变,有报道称,严重的胶质增生和大量的铁沉积可致TIWI 上双侧苍白球出现低信号改变[13]。
本组2 例患儿,例2 全外显子基因检测显示为PANK2基因纯合变异,而其母亲为杂合变异,父亲则未检测到该变异,该结果已经过单基因遗传病位点验证。出现这种基因检测的结果可能原因有:①患儿自身新发变异位点同患儿母亲遗传位点;②患儿父亲实际可能存在此位点变异,但变异深度不够,目前测序水平尚未能够测出;③存在单亲二倍体现象,PANK2基因固定于20p13,若该染色体或该染色体的一个区域全部来源于患儿母亲,该来源恰好包含母源突变,就会出现患儿父亲未检测到该变异、但患儿为纯合变异现象。
PANK 2基因变异与脑组织特殊区域铁沉积之间的联系目前尚不十分清楚。线粒体代谢的改变和线粒体脂类代谢可能的特殊代谢途径,可能是NBIA 神经变性过程的病理基础[14]。目前尚缺乏对NBIA的有效治疗方法。曾经有学者试图通过铁螯合作用清除脑组织中沉积的铁离子,然而既往研究业已显示,此法既未能减少铁离子的沉积,亦对临床病程毫无改善作用[15]。本组例1患儿曾行安坦、左旋多巴药物治疗,治疗1周左右肌张力障碍表现稍有缓解,但2周后再次出现肌张力障碍危象,表现为双上肢屈曲旋转,双手握拳,双下肢向后伸展,呈强直状,腰背呈反折状,喉口发出声音,痛苦面容,可见安坦、左旋多巴对于患儿的治疗效果欠佳。目前临床上对于NBIA 的治疗研究主要集中于PKAN,其余类型,如由于铜蓝蛋白基因变异导致的遗传性铜蓝蛋白缺乏症,以及铁蛋白轻链基因变异导致的神经铁蛋白病的治疗研究尚少。
综上所述,本组2例NBIA患儿经基因测序,发现PANK2基因的可能致病变异,C.515-527del、C.644-645delGAinsAT、c.397A>G(p.Met133Val),为遗传咨询和产前诊断提供了依据,同时也扩展了人类基因组突变数据库。
猜你喜欢
中国典型病例大全(2021年11期)2021-11-02 05:53:51
种子(2021年3期)2021-04-12 01:42:22
中国现代神经疾病杂志(2017年1期)2017-03-29 06:39:34
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
广东药科大学学报(2016年6期)2016-03-10 07:33:32
放射学实践(2015年2期)2015-02-14 05:38:58
中国神经免疫学和神经病学杂志(2014年5期)2014-05-08 06:17:25
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
中国乡村医药(2012年7期)2012-01-22 04:25:49
