
一个中国Usher综合征2型家系中USH2A基因的2个新的致病突变
2020-06-26 09:08何晨昊刘馨屿钟子琳陈建军
同济大学学报(医学版) 2020年3期
何晨昊, 刘馨屿, 钟子琳, 陈建军,3
(1. 同济大学医学院,上海 200092; 2. 同济大学附属第四人民医院脑功能与人工智能转化研究所,上海 200081;3. 同济大学附属第十人民医院儿科,上海 200072)
Usher综合征(Usher syndrome, USH)是一种具有临床和遗传上异质性的常染色体隐性遗传疾病。USH的特征是视网膜色素变性(retinitis pigmentosa, RP),双侧感觉神经性听力障碍和完整的前庭反应[1]。USH是人类先天性视力和听力障碍最普遍的原因。在全球范围内,USH的患病率约(3.3~6.4)/10万[2],目前为止,尚无治疗USH的有效疗法。
在临床上,根据患者视力和听力丧失的严重程度和进展情况,USH分为Ⅰ类USH(USH1),Ⅱ类USH(USH2)和Ⅲ类USH(USH3)[3]。此外,大约20%~30%的病例为非典型性USH。USH1是这3种类型中最严重的一类,USH1患者患有先天性深度听力丧失并一出生就失去视力。不同于USH1患者的先天性耳聋和失明,USH2患者在20岁左右才表现出轻-中度的听力和视力丧失。USH2是USH中最常见的一类,占所有USH患者总数的一半以上[2,4]。USH3患者并非天生耳聋,通常是随着年龄的增长逐渐丧失听力和视力。
到目前为止,已鉴定出16个可能导致USH的基因(https:∥sph.uth.edu/retnet/sum-dis.htm),其中3个基因[USH2A(Usherin)[5],ADGRV1(adhesion G protein-coupled receptor V1)[6]和DFNB31(autosomal recessive deafness 31[7])]是USH2的致病基因。USH2A基因是USH2的主要致病基因,占所有USH2病例的74%以上[8]。该基因位于染色体1q41上,具有2种不同剪接的亚型。1998年,较短的USH2A亚型a首次被鉴定出来[5],而van Wijk等[9]于2004年鉴定出更长的USH2A亚型b。迄今为止,已对USH2A亚型b的全部72个外显子进行了大量的突变分析,发现了许多致病性突变(包括剪接位点处的剪接突变)[10-11]。USH2A亚型b编码有5202个氨基酸的蛋白Usherin,它锚定在细胞膜上[12],并特异性表达在哺乳动物的感光细胞的连接纤毛中,参与从内部到外部的物质运输过程[13]。先前的研究表明,USH2A的突变可能导致非综合征性隐性RP[14-15]。此外,USH2A基因也与触觉敏感性和敏锐度有关[16]。
1 资料与方法
1.1 临床样本收集
本研究选择了8例就诊于同济大学附属第十人民医院的中国患者,经仔细的临床检查,根据患者的光学相干断层扫描(optical coherence tomography, OCT)和视网膜电图(electroretinogram, ERG)结果,以及典型的RP眼底外观,完整的前庭功能和感觉神经性听力障碍诊断为USH2。来自NCBI的USH2A基因参考序列被用作对照。本研究符合《赫尔辛基宣言》所规定的标准,并获得了同济大学医学院附属同济眼科研究所(中国上海)机构审查委员会(IRB)的批准。所有参与者均签署知情同意书。
1.2 样品制备和突变筛选
将所有患者及其亲属的外周血样品收集在EDTA管中。根据制造商的说明,使用RelaxGene血液DNA系统的标准规程(TianGen,中国北京)提取基因组DNA。DNA样品在使用前保存在-80℃的环境中。使用Primer3软件(http:∥primer3.sourceforge.net/)设计了USH2A外显子2至72(包括内含子-外显子边界)的特异性引物。通过聚合酶链反应(PCR)扩增目的片段,PCR产物送去生工生物工程(上海)股份有限公司测序,之后利用Snapgene软件将测序结果与已发表的USH2A基因的DNA序列(NCBI参考序列: NM_206933.3)进行比较,检测有无碱基突变的情况。编号+1的cDNA对应于USH2A的ATG翻译起始密码子中的A。
1.3 错义突变异致病性预测及分析
使用了几种不同的计算机软件,如SIFT和PROVEAN(http:∥provean.jcvi.org/genome_submit_2.php)、PolyPhen-2(http:∥genetics.bwh.harvard.edu/pph2/)和MutationTaster(http:∥www.mutationtaster.org/)来预测错义变体的致病作用。使用Clustal Omega(https:∥www.ebi.ac.uk/Tools/msa/clustalo/),通过将直系同源USH2A蛋白序列(包括小鼠、穴居人、牛、鸡、猕猴和斑马鱼)与人类USH2A蛋白序列进行比对,评估物种间的进化保守性。
2 结 果
2.1 USH2患者的临床特征
该患者年龄为20周岁,性别男,其眼底照片显示了视网膜中不规则的色素团块和视网膜血管的减少等典型的RP特征(图1A),患者的OCT成像表明其视网膜厚度明显减少(图1B)。此外,该患者有中等程度的听力障碍,纯音听力图测试分析显示其双侧导气和骨导听觉下降,鼓室图显示为As型,这意味着中耳传播系统活动受到限制。无法检测到患者的ERG波振幅。这些特征支持了USH2的诊断。
2.2 USH2A突变的致病性分析
本研究将8例USH2患者的USH2A基因外显子测序结果与已发表的USH2A基因的DNA标准序列进行了比对,并与SNP数据库进行比对分析。最终在其中1例USH2患者身上,发现了2个新的杂合突变。根据计算机算法的结果,预测这2个新的杂合突变是致病性的,见表1。此外,在该患者身上,本研究还发现了其他6个已报道的被预测为非致病性的变化,见表2。对该患者的其他2个USH2致病基因ADGRV1和DFNB31的检测结果显示未发现有致病突变存在。

图1 USH2患者的临床检查图像Fig.1 Fundus images of the USH2 patientA: 该患者左眼眼底图;B: 该患者右眼眼底图;C: 该患者左眼的OCT图像
这2个新的杂合突变分别是位于外显子2上的移码突变c.230dupA(Phe78Valfs*30)(图2A)和位于外显子19上的错义突变c.4085C>T(p.Pro1362Leu)(图2B)。在该患者的母亲身上,本研究也发现了错义突变c.4085C>T(p.Pro1362Leu),但却没有发现移码突变c.230dupA(Phe78Valfs*30),在该患者的父亲身上未发现有致病突变。该患者的父母都是表型正常的正常人,家系图显示该患者的遗传遵循常染色体隐性遗传方式,见图3。
此外,本研究确定了这2个致病突变在usherin蛋白上的定位,见图4A,对于错义突变c.4085C>T(p.Pro1362Leu),使用Clustal Omega软件分析比对不同物种(包括人类,穴居人,小鼠,猕猴,鸡,斑马鱼和牛)之间USH2A序列,确定了其氨基酸序列具有高度的保守性,见图4B。

表1 本研究中鉴定出的USH2A基因致病突变及其分析软件预测结果Tab.1 Identified pathogenic variants in USH2A gene in this study and their prediction results from the analysis programs
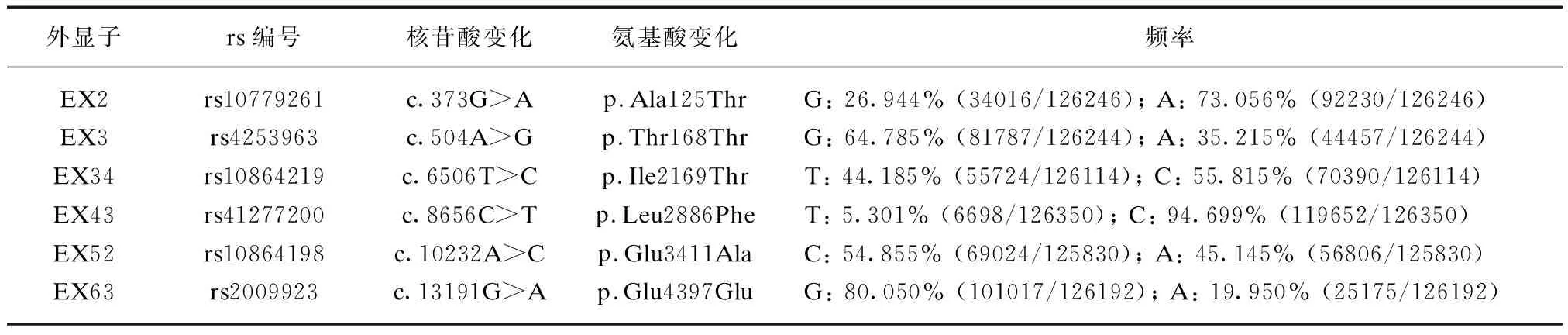
表2 本研究中鉴定出的非致病性突变Tab.2 Variants predicted non-pathogenic in this study
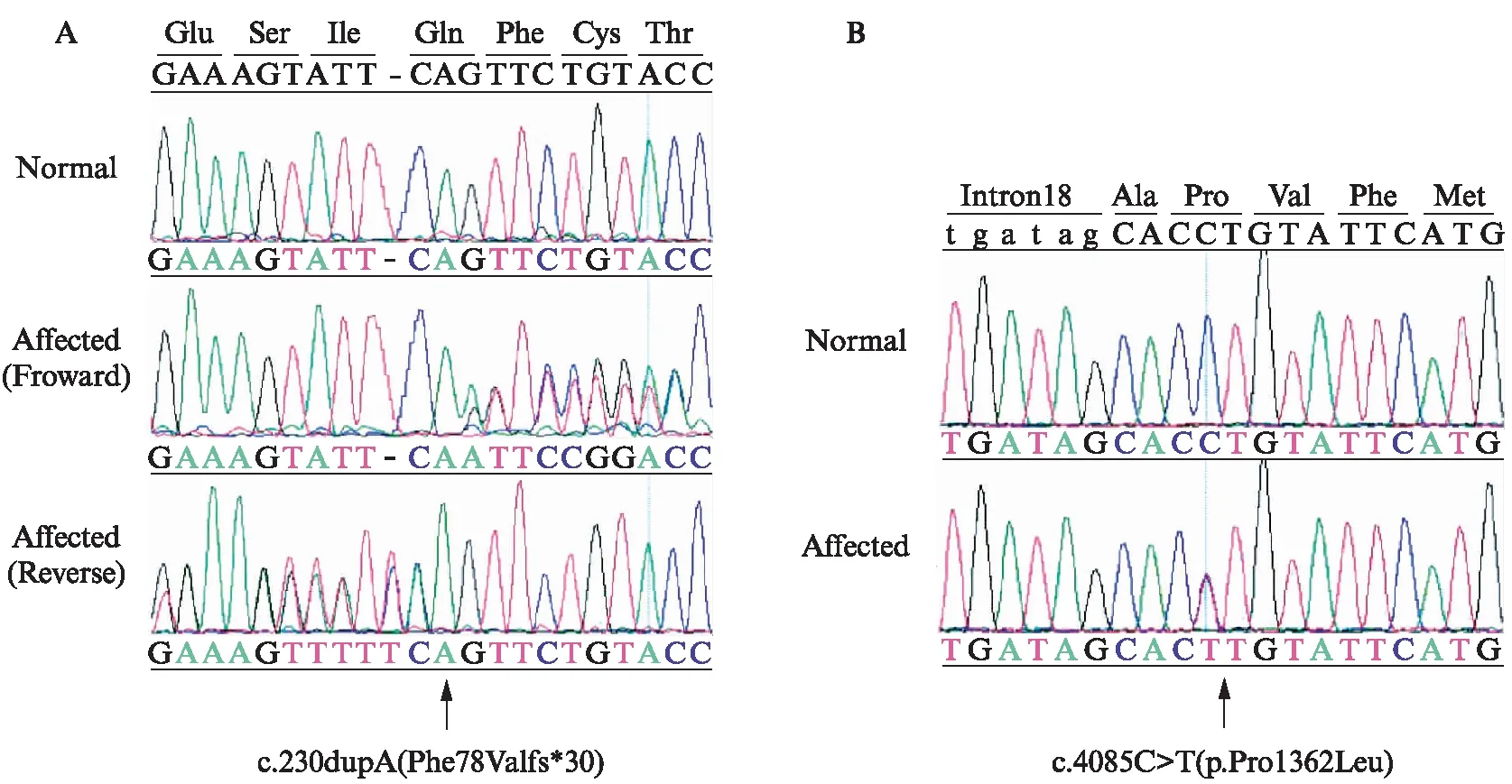
图2 本研究中鉴定的新的USH2A基因致病突变的测序分析结果Fig.2 Direct sequencing analysis of the novel pathogenic variantsin USH2A gene identified in this studyA: 新的移码突变c.230dupA(Phe78Valfs*30)及其对应的野生型序列;B: 新的错义突变c.4085C>T(p.Pro1362Leu)及其对应的野生型序列
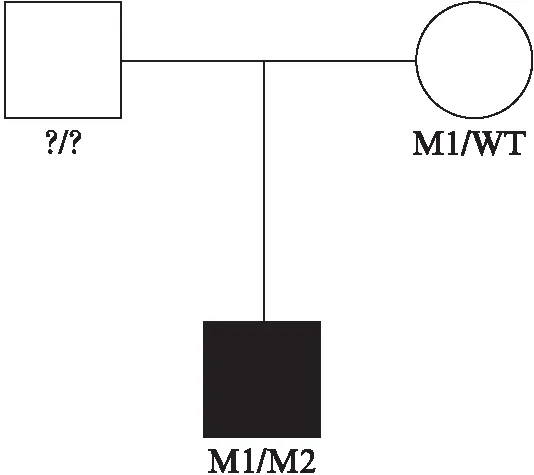
图3 USH2患者的家系图Fig.3 Pedigree of a Chinese USH2 patient’s family黑色代表患有USH2,白色为正常人;正方形表示男性,圆圈表示女性;M1: 新的移码突变c.230dupA(Phe78Valfs*30);M2: 新的错义突变c.4085C>T(p.Pro1362Leu);WT: 野生型
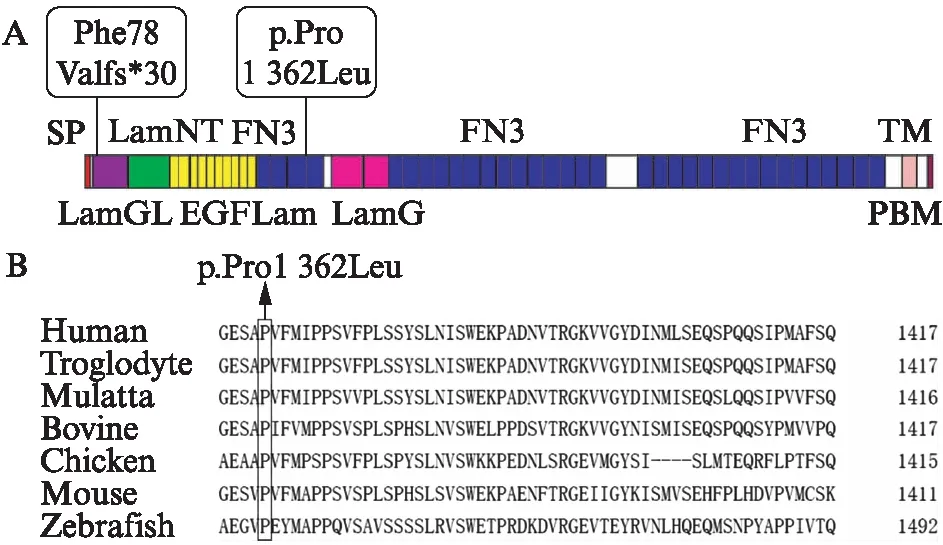
图4 致病突变的定位及氨基酸序列比对Fig.4 Location of pathogenic mutation and amino acid sequence alignmentA: 本研究中鉴定出的新的外显子突变在USH2A亚型b上的定位;B: Clustal Omega软件的氨基酸序列比对情况
3 讨 论
目前,人们已经鉴定出了与USH相关的16个基因,其中3个是引起USH2的基因。在这3个基因中,USH2A可导致USH2病例的30%~40%和隐性RP病例的10%~15%[17]。Usherin定位于哺乳动物感光细胞中空间受限的膜微区[13]。先前的研究表明,先天性usherin蛋白突变可以诱导纤毛连接障碍,并最终导致失明[18]。
到目前为止,在中国患者中进行的USH2A突变筛查已经发现了25个致病突变[15,19-23]。在中国南方人群中,8.47%的散发性RP患者属于USH[19]。本课题组在1例被诊断为USH2的中国患者的USH2A基因中鉴定了2个新的杂合致病突变(1个移码突变和1个错义突变),并发现了其他6个已报道的非致病性的变化。
USH2A亚型b包含8个结构域,包括N端的信号肽(signal peptide, SP),层粘连蛋白G样结构域(laminin G-like domain, Lam GL),层粘连蛋白N端(laminin N-terminal, Lam NT),层粘连蛋白型EGF样结构域(laminin-type EGF-like domain, EGF Lam),Ⅲ型纤连蛋白(fibronectin Type Ⅲ, FN3),层粘连蛋白G结构域(laminin G domain, Lam G),跨膜区(transmembrane region, TM)和位于其C末端的PDZ结合模体(PDZ-binding motif, PBM)[9]。Usherin通过PBM结构域与harmonin和whirlin两种支架蛋白的PDZ结构域相互作用整合到USH蛋白网络中[24]。新的移码突变c.230dupA(Phe78Valfs*30)在外显子2上,定位于Lam G结构域,导致77个密码子移码并影响下游的85个氨基酸,可能导致usherin蛋白质结构发生重大变化。而新的错义突变c.4085C>T(p.Pro1362Leu)发生在多个物种中高度保守的位置上,使亮氨酸代替原来的脯氨酸,定位于第3个FN3结构域。
在所有3个USH2致病基因中,USH2A基因是最重要的致病基因,由USH2A编码的usherin蛋白对哺乳动物感光细胞的长期维持至关重要[13]。因此,鉴定USH2A基因中的致病突变不仅有助于阐明USH2A在USH2中的作用,而且对USH2的临床诊断及寻找其有效的治疗方法具有重大的意义。这些致病突变导致视觉缺陷和听力障碍的具体原因目前尚未被阐明,有待进一步的功能和机制上的研究。
总之,本研究通过全外显子测序鉴定了可能导致2型Usher综合征的2个新的杂合USH2A基因致病突变。这一结果扩大了已知的USH2A基因致病突变的范围。
猜你喜欢
安徽农业大学学报(2022年2期)2022-11-09
电子科技大学学报(2022年5期)2022-10-29
广西医科大学学报(2022年5期)2022-06-07
中国循证儿科杂志(2022年2期)2022-05-26
昆明医科大学学报(2022年3期)2022-04-19
中国土壤与肥料(2021年5期)2021-12-02
中国生殖健康(2020年4期)2021-01-18
中南医学科学杂志(2019年6期)2019-12-05
肿瘤预防与治疗(2019年6期)2019-07-30
中国动物保健(2015年4期)2015-10-21
