
丁苯树脂增韧改性含硅芳炔树脂的性能
2020-06-18 08:16马峰岭李小慧金石磊段家真
理化检验(物理分册) 2020年6期
马峰岭, 李小慧, 金石磊, 段家真, 韩 瑞
(1. 上海材料研究所 上海市工程材料应用与评价重点实验室, 上海 200437;2. 上海安缔诺科技有限公司, 上海 200437)
含硅芳炔树脂(PSA,结构式见图1)是通过在芳基乙炔树脂主链结构上引入硅元素得到一种无机有机杂化树脂,具有优良的耐高温性能、工艺性能和介电性能,主要应用于航空、航天、军工等领域。但是含硅芳炔树脂固化后会形成高交联密度的网络结构,使得树脂质脆,后期机械加工过程中很容易发生脆裂或脆断。机械加工工艺性差,限制了PSA的广泛应用[1]。同时,由于PSA固化物仍会残留少量的未固化炔键,导致PSA在高频率电场下介电性能下降[1],影响了其在电子信息领域的应用,特别是在高频通信领域[2]。
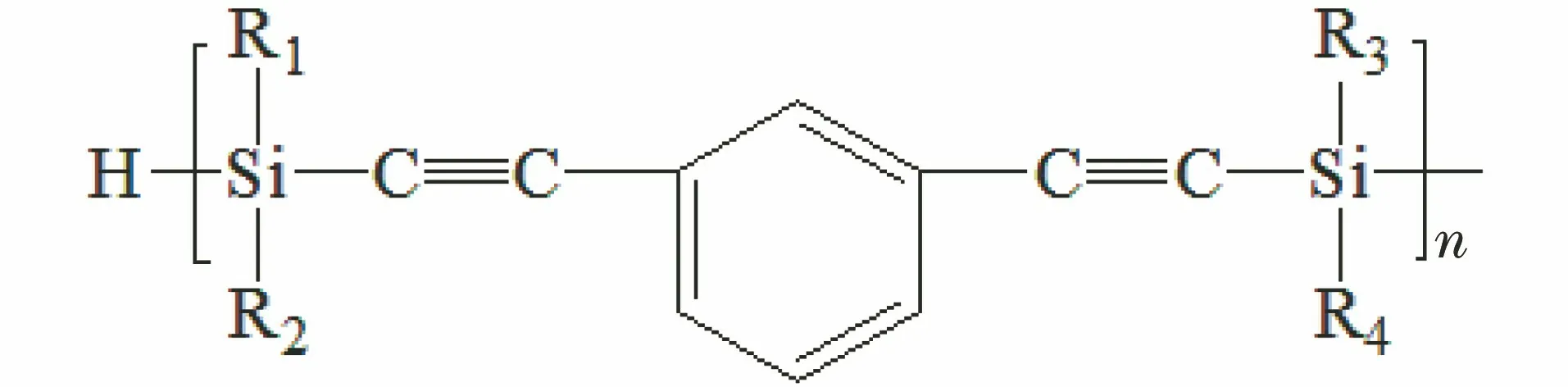
图1 含硅芳炔树脂分子结构式Fig.1 Molecular structure of PSA
国内外针对PSA力学性能较差的问题开展了大量的研究,其中较多采用共混改性的方法增韧改性PSA[3-9]。汤乐旻等[3]采用炔醚及噁嗪化合物改性PSA,所制备的模压成型复合材料弯曲强度提高了53.1%。李晓杰等[4]合成了端乙炔基聚醚酰亚胺并与PSA共混得到改性树脂,较好地提高了树脂的弯曲强度。目前,由于同类型耐高温树脂种类较少,采用其他耐高温树脂增韧改性PSA的研究也受到限制,阻碍了PSA的发展应用。
丁苯树脂(SBR,结构式见图2)是一种热固性非极性聚合物,其侧基含有乙烯基基团,具有交联反应活性,另外含有刚性侧基基团苯环,树脂分子链结构呈柔性伸展且极性较弱,具有优异的介电性能和加工性[10-11]。笔者采用SBR共混改性PSA,并以双叔丁基过氧化二异丙基苯(BIBP)作为促进剂,对SBR/PSA浇注体的结构与性能进行了研究。

图2 丁苯树脂分子结构式Fig.2 Molecular structure of SBR
1 试样制备与试验方法
1.1 试验材料
试验材料为华东理工大学研发的PSA,克雷威利化工有限公司的SBR,阿科玛投资有限公司的BIBP。
1.2 试验设备
主要试验设备为:东莞市鼎麓机械设备有限公司生产的型号为RAY-NJ01的凝胶化时间测试仪;PerkinElmer®铂金埃尔默公司生产的型号为DSC-6的示差扫描量热法仪(DSC);TA仪器公司生产的型号为TGA-550的热失重分析仪(TGA);PerkinElmer®铂金埃尔默公司生产的型号为PE-1 000的傅里叶变换红外光谱仪(FTIR);上海宙山精密仪器有限公司生产的型号为zmm-500的金相显微镜;PHILIPS公司生产的型号为 XL30的扫描电子显微镜(SEM);德国QWED公司生产的型号为Q-Meter的微波介质谐振器;美特斯工业系统(中国)有限公司生产的型号为MTS CMT5305的万能力学试验机。
1.3 试样制备
采用熔融共混的方法,按照不同配比,将SBR加入到PSA中,需要同时加入一定比例的促进剂,然后在80 ℃下搅拌30 min,得到棕黄色SBR/PSA混合液,各试样成分配比如表1所示。

表1 各试样成分配比(质量分数)Tab. 1 Composition ratio of samples (mass fraction) %
1.4 试验方法
将上述搅拌均匀的不同配比的树脂混合液置于真空烘箱内,在80 ℃和-0.1 MPa下真空脱泡30 min,然后倒入模具当中,放入高温烘箱固化。固化工艺为:150 ℃保温2 h→180 ℃保温2 h→220 ℃保温2 h→250 ℃保温2 h。然后制备出尺寸为50 mm×15 mm×4 mm的弯曲性能试样和尺寸为50 mm×15 mm×4 mm的介电性能试样。
采用凝胶时间测试仪,取200 mg不同配比混合树脂测定171 ℃下平板凝胶时间;采用DSC进行热分析(氮气气氛,升温速率 10 ℃·min-1,25~350 ℃);采用TGA进行热失重分析(氮气气氛,升温速率20 ℃·min-1,25~1 000 ℃);采用微波介质谐振器测试试样在5 GHz频率下的介电性能;采用万能试验机进行弯曲试验,试验速度为2 mm·min-1;采用扫描电镜和金相显微镜观察试样的断面形貌。
2 结果与讨论
2.1 SBR/PSA树脂固化行为
图3为不同比例SBR/PSA树脂体系的DSC曲线。由图示可知,PSA树脂有一个较强放热峰,起始反应温度为170 ℃,固化反应峰值温度为220 ℃,固化反应结束温度为260 ℃;SBR也有一个放热峰,放热量比PSA少很多,且反应温度高于PSA,起始反应温度达210 ℃,说明SBR热反应活性低于PSA。这是因为SBR具有较少的乙烯基反应基团,且乙烯基热引发交联的反应活性较低,使得SBR在无促进剂的条件下,不容易发生热交联反应。分析30SBR/PSA热分析曲线可知,其反应放热量大大减少,反应温度升高,说明在无促进剂的条件下SBR的加入降低了PSA体系的反应活性;对比30SBR/PSA/B可知,其反应放热量增加,反应温度降低,促进剂BIBP的加入提高了反应活性。对于SBR/PSA/B体系,存在两个放热峰,其中一个放热峰峰值温度为190 ℃,且随着SBR含量的增加,反应放热量逐渐减少,反应活性降低,反应起始和结束温度无明显变化;另一个放热峰峰值温度为220 ℃,放热量很少,随着SBR含量的增加,该反应放热峰变的越来越弱,这主要是受到PSA树脂炔基反应所造成的,表现为PSA剩余少量内炔基基团在高温条件下的反应放热。

图3 各试样的DSC曲线Fig.3 DSC curves of samples
2.2 SBR/PSA树脂流变性能

表2 各试样的凝胶时间Tab.2 The gelation time of samples
表2为不同SBR/PSA树脂体系在171 ℃下的凝胶时间。由表2可以看出,在无促进剂的条件下,SBR/PSA体系比PSA的凝胶时间更长,再次说明SBR树脂的加入降低了PSA的反应活性。BIBP促进剂的加入,使得30SBR/PSA的凝胶时间由原来的1 600 s缩短为170 s,大大提高了反应活性。同时,随着SBR含量的不断增加,PSA凝胶时间逐渐增加,这是因为SBR的加入降低了PSA的反应活性,与DSC热分析曲线表征结果相一致。
2.3 SBR/PSA树脂固化物红外光谱
图4为不同配方SBR/PSA树脂固化物的红外光谱分析图。由图可知,SBR/PSA树脂均在2 154 cm-1处出现了-C≡C-内炔基基团的特征峰,而在3 291 cm-1处未见PSA端基-C≡CH基团的特征峰,也未见SBR的端基-C=H2乙烯基特征峰,表明SBR/PSA树脂按照上述固化工艺可以保证固化反应完全。
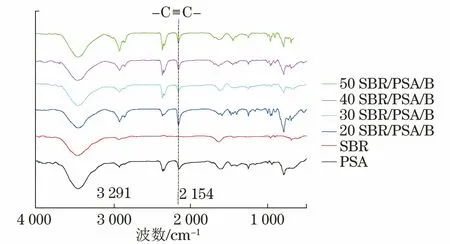
图4 各试样的FTIR曲线Fig.4 FTIR curves of samples
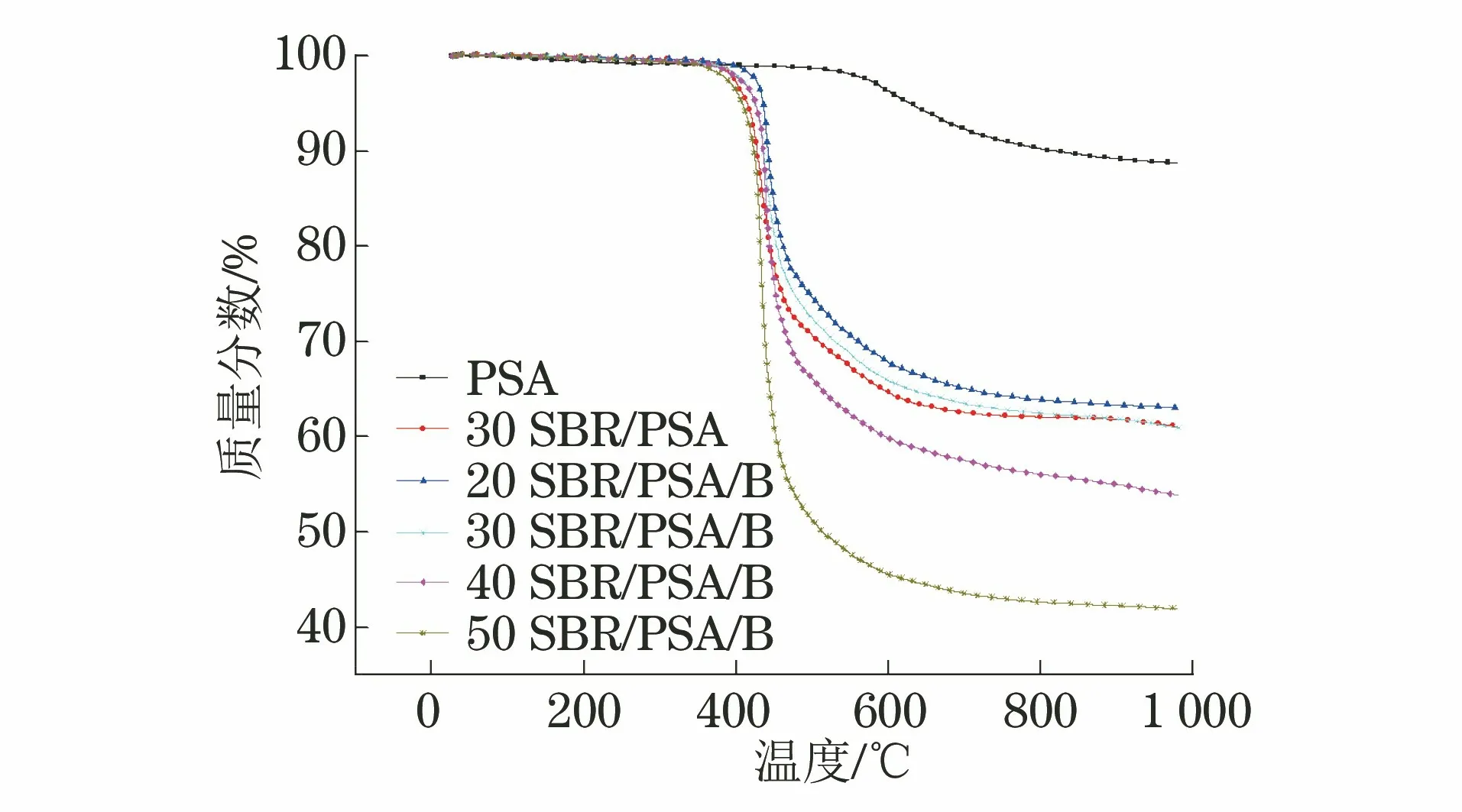
图5 各试样的TGA曲线Fig.5 TGA curves of samples
2.4 SBR/PSA树脂固化物热稳定性
图5为不同SBR/PSA配方体系树脂固化物的TGA曲线图。由图5可以看出,PSA具有极高的耐高温性能,失重5%时的温度可达630 ℃,1 000 ℃时的残碳量为89%。随着SBR的加入,SBR/PSA树脂耐高温性能明显降低,但是在400 ℃以下仍保持较好的热稳定性。随着温度进一步升高,SBR/PSA树脂开始发生热分解失重,热失重主要发生在400~600 ℃。其中SBR含量为20%,30%,40%,50%时,失重5%时的温度分别为435,425,420,408,1 000 ℃,热解残留率分别为63%,61%,54%,42%。SBR的加入导致热失重增加的主要原因在于,所用SBR分子链主链结构含有耐热温度较低的C-C直链结构,其耐热分解能力低于有大量环状结构的PSA,所以SBR加入越多,SBR/PSA树脂的热稳定性能下降越明显。
2.5 SBR/PSA树脂介电性能
图6为不同含量SBR改性PSA浇注体的介电性能变化曲线。由图6可以发现,随着SBR加入,SBR/PSA树脂介电常数和介电损耗均随之降低,介电性能得到明显改善,且随着SBR含量的不断增加,介电常数和介电损耗持续下降,当SBR含量增加到50%时,其介电常数和介电损耗分别降低至2.58,0.002 9,低于PSA树脂的2.89,0.003 6。这主要是因为PSA含有大量的芳炔基团,当其固化形成密实的网络结构时,仍残留少量具有一定极性的内炔基基团;而SBR为非极性聚合物,其经交联固化后形成稳定的网状结构,在电场作用下很难发生极化并对电磁场产生影响,具有极低的介电常数和介电损耗,因此SBR的加入可以有效提高PSA的介电性能。

图6 SBR加入量对试样介电性能的影响Fig.6 Effect of the amount of SBR on the dielectric properties of samples
2.6 SBR/PSA树脂弯曲性能
图7为不同含量SBR改性PSA浇注体的弯曲性能变化曲线。由图7可知,随着SBR含量的增加,SBR/PSA树脂的弯曲强度和弯曲模量均呈先增加后减小的变化趋势。这是因为PSA分子结构为多网状结构,分子链运动能力很弱,树脂固化物呈脆性断裂,而SBR分子链结构含有较多柔性C-C直链,SBR的加入可以提高PSA的结构韧性,当SBR含量为40%时,SBR/PSA树脂的弯曲强度和弯曲模量分别增加了93%和173%;但是因SBR具有较少的反应活性乙烯基基团,当SBR含量高于40%时,SBR/PSA树脂固化物分子结构交联度大大降低,导致其弯曲性能大幅下降。

图7 SBR加入量对试样弯曲性能的影响Fig.7 Effect of the amount of SBR on the bending properties of samples
2.7 SBR/PSA树脂断面形貌
图8和图9分别为30SBR/PSA和30SBR/PSA/B浇注体断面的光学显微图像。如图8和图9所示,无促进剂的30SBR/PSA树脂体系(图8)发生明显的结构相分离,白色的区域为SBR,颜色较深的区域为PSA,而30SBR/PSA/B树脂浇注体断面呈均相结构(图9),说明BIBP促进剂的加入有利于SBR/PSA树脂形成均相结构。结合上述热分析结果可知,PSA和SBR反应活性相差较大,PSA起始反应温度为170 ℃,SBR起始反应温度为210 ℃,SBR/PSA树脂在较低温度首先发生PSA固化反应,当PSA反应加剧时,SBR才刚开始发生反应,反应活性的巨大差异导致PSA与SBR无法进行共固化交联而形成均一的网络结构。

图8 30SBR/PSA断面形貌Fig.8 Morphology of30SBR/PSA fracture surface

图9 30SBR/PSA/B断面形貌Fig.9 Morphology of 30SBR/PSA/B fracture surface
图10~图13为SBR/PSA树脂浇注体断面的SEM形貌。图10为 20SBR/PSA/B树脂的SEM形貌,由图10可知,断面结构平整,无裂纹产生,呈典型的脆性断裂,表明20%的SBR增韧改性PSA的效果较弱。由图11和图12可知,随着SBR含量继续增加,SBR/PSA改性树脂断面结构出现明显的拔出裂纹,表明增韧效果明显;但是当SBR含量增加至50%时,如图13所示,SBR/PSA树脂断面呈细小丝状拔出裂纹,无法形成强度较高的铆钉断裂结构,断裂强度减小,与弯曲性能的表征结果保持一致。

图10 20SBR/PSA/B断面的SEM形貌Fig.10 SEM morphology of 20SBR/PSA/B fracture surface

图11 30SBR/PSA/B断面的SEM形貌Fig.11 SEM morphology of 30SBR/PSA/B fracture surface

图12 40SBR/PSA/B断面的SEM形貌Fig.12 SEM morphology of 40SBR/PSA/B fracture surface

图13 50SBR/PSA/B断面的SEM形貌Fig.13 SEM morphology of 50SBR/PSA/B fracture surface
3 结论
(1) SBR反应活性较低,反应温度较高,与PSA固化后无法形成均相体系,在BIBP促进作用下,SBR反应温度降低,可与PSA在同一温度条件下发生共交联固化反应,并形成均相体系。
(2) SBR具有优良的力学性能,当SBR含量为40%(质量分数)时,SBR/PSA弯曲强度提高了93%,弯曲模量提高了173%,改善了PSA的韧性,为最佳状态;当SBR树脂含量继续增加时,SBR/PSA树脂交联密度大大降低,力学性能开始降低,材料断裂界面微观结构由较大的拔出裂纹形态转变为细小的丝状裂纹。
(3) SBR作为非极性聚合物,介电性能优异,当SBR含量为40%(质量分数)时,介电常数和介电损耗分别降低至2.58和0.003。
猜你喜欢
阅读(快乐英语高年级)(2021年11期)2021-03-08
潍坊学院学报(2020年2期)2021-01-18
陶瓷学报(2019年6期)2019-10-27
中国盐业(2018年18期)2019-01-14
电子制作(2018年10期)2018-08-04
橡胶科技(2018年4期)2018-02-17
电子制作(2017年7期)2017-06-05
汽车零部件(2015年1期)2015-12-05
橡胶工业(2015年5期)2015-08-29
橡胶工业(2015年11期)2015-08-01
