
全固态钠离子电池硫系化合物电解质
2020-06-18 11:52:12陈光海白莹高永晟吴锋吴川
物理化学学报 2020年5期
陈光海,白莹,高永晟,吴锋,吴川
北京理工大学材料学院,环境科学与工程北京市重点实验室,北京 100081
1 引言
为缓解能源危机和减少温室气体的排放,大力开发太阳能、风能、潮汐能等可再生清洁能源是人类生存和发展做出的重要选择。而将间歇性的可再生清洁能源进行规模化存储与高效转换需要先进的技术。作为一种先进的能量存储与转换技术,二次电池体积小、能量转换效率高、维护简单,已经在移动电子和纯电动/插电混合汽车等领域得到了广泛应用1。可充电钠离子电池与锂离子电池具有类似的电化学性质且钠资源丰富、分布广泛、成本低,在大规模储能领域,钠离子电池比锂离子电池更具竞争力2,3。不同于功率型电动汽车用的动力电池,低成本、高安全性、长循环寿命是可充电电池用于大规模储能的基本要求。近年来,锂离子电池安全事故频发,这与有机液态电解质易燃、易漏、易导致枝晶生长等本质特征密切相关。用固态电解质替换液态电解质原则上可以消除上述安全隐患。因此,全固态钠离子电池是一种极具有发展前景的大规模储能技术之一。
固态电解质是全固态钠电池的核心组成部分,不仅要隔离正负极以防短路,而且要传导钠离子。根据组成不同,钠离子固体电解质可分为无机固体电解质、聚合物电解质4,5和复合电解质三大类。无机固体电解质包括氧化物Na-β-Al2O3、钠超离子导体NASICON、硫系化合物和硼氢化物。陶瓷基氧化物电解质的电化学窗口宽、热稳定性好、离子电导率高、离子迁移数高、抗冲击和振动能力强6,7,然而刚性强、易碎。同时,陶瓷制备过程需要高温热处理来减小晶界阻抗,与电极材料共烧结来减小电解质与电极界面接触电阻7。一方面需要高能耗,不够经济环保;另一方面,许多电极材料在高温下与电解质会发生反应,电极材料失效或者界面生成离子绝缘层导致电池性能差8,9,因此陶瓷基固态钠离子电池电极材料可选范围很窄。
硫系化合物固态电解质可变形,能够通过简单的冷压使电解质与电极界面紧密接触,减小晶界和界面阻抗10,11。此外,硫系化合物固态电解质电导率高,Li7P3S11(17 mS·cm-1)12、Li10GeP2S12(12 mS·cm-1)13等几种先进的锂离子硫化物固体电解质的离子电导率与液态电解质相当。对于制备块状固态电池,在各种固态电解质中,具有良好成型性的硫系化合物固态电解质是一种不错的选择,在大规模储能用固态钠离子电池领域具有重要的应用价值。
本文系统地总结了钠离子硫系化合物固态电解质的研究进展,从晶体结构出发综述钠离子传导机制及增强电导率的方法,并深入探讨了硫系化合物电解质的化学稳定性、电化学稳定性、与电极的界面稳定性,最后介绍了硫系化合物电解质基全固态钠离子电池的发展现状并分析了其存在的挑战与解决方案。
2 钠离子硫系化合物固态电解质的结构与性质
2.1 Na3MS4 (M = P, Sb)系列
作为钠离子硫系化合物电解质最典型的代表之一,硫基硫化物固体电解质引起了人们的极大关注。早在1992年,Jansen和Henseler14通过固相法合成了四方相单晶Na3PS4,50 °C下离子电导率为4.17 × 10-6S·cm-1,510 °C高温下离子电导率达8.51 × 10-2S·cm-1,钠离子在晶格空腔内呈直线型和“Z”字形排列,在高温下钠离子各向同性高速迁移。2012年,Hayashi等15首次提出了室温离子电导率高达2 × 10-4S·cm-1的立方相Na3PS4玻璃、玻璃-陶瓷电解质,这一具有里程碑意义的工作掀起了人们对钠离子硫化物固体电解质的研究热潮。
Na3PS4有两种不同的晶相,立方相c-Na3PS4和四方相t-Na3PS4,c-Na3PS4是高温下的稳定相,t-Na3PS4则是低温下的稳定相。两种不同晶相Na3PS4的晶体结构如图1所示,t-Na3PS4比c-Na3PS4两者晶格参数最大差异为1%,体积差异0.2%。最重要的差异是钠的占位不同,c-Na3PS4有两种不同的Na对称位点,部分占据的6b位点(Na1,占据百分率0.8)和部分占据的12d位点(Na2,占据百分率0.1)16;在t-Na3PS4中,可看作是c-Na3PS4的Na1(6b)位点裂分为4d位置的Na1位点和2a位置的Na2位点,4d位点相对c-Na3PS4的位置移动0.05 nm17,18。经过轻微旋转、钠离子再分布就能实现c-Na3PS4向t-Na3PS4转化。
Na2S与P2S5以摩尔比75 : 25混合球磨可以制得完全无定形的玻璃态Na3PS4;经过270 °C热处理,玻璃粉末再结晶得到立方相玻璃-陶瓷态Na3PS4;如果经过420 °C热处理,得到的是四方相玻璃-陶瓷态Na3PS4(图2a)。参与固相球磨反应的Na2S与P2S5的摩尔比影响Na3PS4的局部结构、热力学行为和电化学导电性,当Na2S摩尔分数占80%时,玻璃态的Na3PS4室温离子电导率最高19。进一步细化热处理温度梯度,发现加热至300 °C以上,立方相Na3PS4就能转化为四方相Na3PS4。除了是否进行热处理,球磨转速、球磨时间、反应物纯度等制备工艺对Na3PS4的纯度、结晶态和晶型也具有重要影响20。当以400 r·min-1转速球磨5 h以上,才能减少反应残留物Na2S;如果使用高纯度(99.1%)的晶态Na2S,玻璃-陶瓷态Na3PS4室温离子电导率能提高一倍(4.6 × 10-4S·cm-1)21。立方相Na3PS4的纯度随球磨时间的变化和立方相Na3PS4随热处理温度变化向四方相Na3PS4变化的过程见图2b,c。总的来说,玻璃-陶瓷态Na3PS4的电导率比玻璃态Na3PS4电导率高,立方相Na3PS4的电导率比四方相的Na3PS4电导率高。分子动力学模拟表明钠离子在c-Na3PS4和t-Na3PS4中的传导是相似的,因此,t-Na3PS4理论上应该也有很高的离子电导率18。Kanno等22发现经过加热-淬火-加热连续处理能够得到晶格体积更大的t-Na3PS4,淬火能够使高温相在室温下稳定并增加钠离子空位,而淬火后的二次加热能够使晶格扩张,进一步增加钠离子空位和间隙,电导率提高了一个数量级(3.39 ×10-3S·cm-1)。
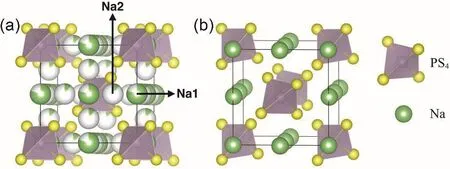
图1 (a)立方相c-Na3PS4 16和(b)四方相t-Na3PS4 17的晶体结构Fig. 1 Crystal structures of (a) c-Na3PS4 16 and (b) t-Na3PS4 17.
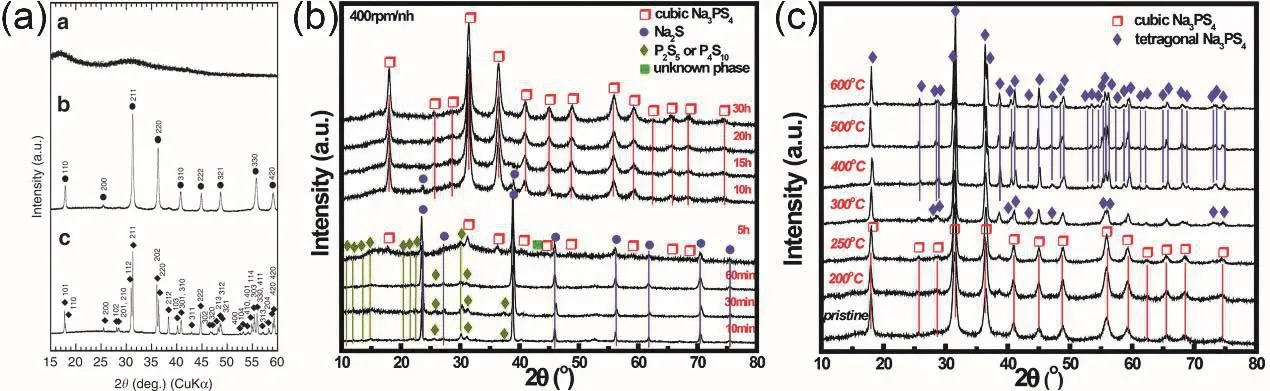
图2 (a)立方相Na3PS4玻璃、经过270 °C热处理的立方相Na3PS4玻璃-陶瓷及经过420 °C热处理的四方相Na3PS4玻璃-陶瓷的X射线衍射图谱15,(b)立方相c-Na3PS4和(c)四方相t-Na3PS4的X射线衍射图谱20Fig. 2 (a) XRD patterns of the cubic Na3PS4 (c-Na3PS4) glass, c-Na3PS4 glass-ceramic sample heated at 270 °C and tetragonal Na3PS4 (t-Na3PS4) glass-ceramic sample heated at 420° C 15. XRD patterns of (b) c-Na3PS4 and (c) t-Na3PS4 20.
目前提高硫化物固体电解质电导率的常见方法主要包括掺杂和二元共混两种。元素掺杂是常用增强电导率的改性方法,对P位进行异价掺杂或对S位进行卤素掺杂均可以提高硫化物固体电解质Na3PS4的电导率。下面对两种掺杂增强电导率的不同机理进行总结。
第一性原理研究表明c-Na3PS4的室温离子电导率较低(1.1 × 10-4mS·cm-1),活化能较高(537 meV),但是,引入钠离子间隙能够使离子电导率提高4个数量级(1.06 mS·cm-1),活化能降至256 meV16。比如,用Sn4+、Ge4+、Ti4+、Si4+等四价离子取代P5+,可以补偿钠离子间隙,得到理想的电导率。理论计算得出掺杂6.25% Sn或Ge的硫化物结构中钠离子通道尺寸最大,Sn掺杂的结构中通道体积比其他结构高4%16。Admas等23用10%的Ge4+、Ti4+、Sn4+对中c-Na3PS4的P位进行掺杂,四价阳离子掺杂会引入更多的Na,因此晶体体积会随掺杂量的增加而增大;晶体体积大小与掺杂离子半径大小呈线性关系,同样掺杂量时,Sn4+掺杂后晶体体积最大。掺杂离子半径越大,钠位点过度拥挤使钠位点能越高,利于降低间隙迁移势垒,并且更有利于使掺杂相稳定。因此,研究者认为Sn掺杂是最有希望提高电导率并得到稳定掺杂相c-Na3.1Sn0.1P0.9S4,与Ong等16的理论计算结果一致。
实验表明,对t-Na3PS4的P位用0.38 mol离子半径更大的合金元素As掺杂后,晶格膨胀和Na-S键长增长提高电导率;然而,As—S键比P—S键弱,导致Na—S键吸引力更强,因此Na—S键键长缩短,掺杂过量的As会使电导率降低。Na3P0.62As0.38S4室温离子电导率高达1.46 mS·cm-1,与有机液态电解质(1–6 mS·cm-1)相当24,25。合金阴离子中这种更长的键长和更弱的键强对电导率的“双刃剑”效应在等合金阴离子中都存在26。
卤素原子引入t-Na3PS4替换S原子(8e位点)形成t-Na3-xPS4-xXx(X = F,Cl,Br,和I)的同时在Na2位点引入钠空位,当掺杂量x为0.0625时,形成能相对较低,且理论计算表明F-掺杂形成能最低,掺杂最容易,其次是Cl-掺杂,I-掺杂,Br-掺杂形成能较高,相对困难17。然而,Wagemaker等18的计算结果表明,从F-、Cl-到Br-,离子半径依次增大,掺杂后的t-Na3PS4离子电导率也逐渐升高,I-掺杂后电导率最低。离子半径大,卤素原子与P原子之间的距离更远,钠离子扩散发生的区间更小,从Na1位点到邻近Na1位点的过渡更不容易发生,因此对提升电导率不利。掺杂不仅引入空位,对微观扩散至关重要;掺杂后声子引起的自由体积大,增加钠离子从一个位点向邻近位点的过渡几率,都有利于提高离子电导率。
往Li3PS4中混入一定量的Li4SiS4得到的二元混合物离子电导率比Li3PS4和Li4SiS4任一单组份的离子电导率都要高27。同样,通过将Na3PS4和Na4SiS4共混的方式引入Si元素也可以提高c-Na3PS4玻璃-陶瓷电解质的电导率28。Na3PS4与玻璃态Na4SiS4以摩尔比94 : 6球磨共混后经过220 °C得到二元混合玻璃-陶瓷电解质94Na3PS4·6Na4SiS4室温离子电导率最高,7.4 ×10-4mS·cm-1。晶体结构精修显示,94Na3PS4·6Na4SiS4在Na2位点的占有率比c-Na3PS4玻璃-陶瓷中高,因此离子电导率更高29。
与Na3PS4类似,Na3SbS4也有四方相Na3SbS4(t-Na3SbS4)和立方相Na3SbS4(c-Na3SbS4)两种不同的相。四方相Na3SbS4(t-Na3SbS4)晶胞中,a=b=0.71597 nm,c= 0.72906 nm,Sb原子占据2b位点,S原子占据8e位点,Sb原子与S原子之间的距离为0.2362 nm30。立方相Na3SbS4晶胞中Na只占据6b位点31,而四方相内Na占据4d位点(Na1)和2a位点(Na2),与c轴平行排列的Na1位点(图3a)和与a或b轴平行以“Z”字形交替排列的Na1、Na2位点(图3b)占据SbS4四面体组成的空隙,两者正交组成三维钠离子传导通道。Na1位点和Na2位点分别与6个S原子和8个S原子配位(图3c)。与面心立方亚晶格具有最低的活化能和最高的离子电导率类似,Na3SbS4中严重扭曲的S原子立方亚晶格也有助于钠离子扩散32。与Na3PS4、Na3PSe4不一样,元素组成比为3 : 1 : 4的四方相Na3SbS4中,Na没有完全占据Na2位点,而是80%Na和20%空位共同占据Na2位点,空位摩尔比例为2.5%,仍然是空位传导的机制,室温离子电导率为1.77 mS·cm-1。
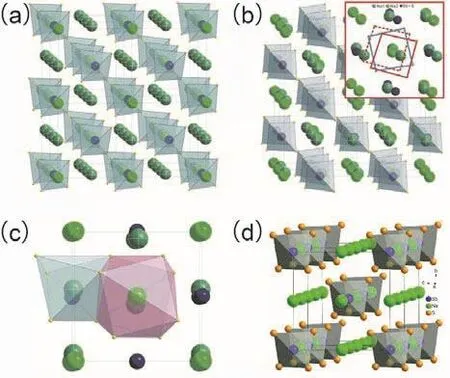
图3 (a) Na3SbS4沿c轴方向晶体结构,(b) Na3SbS4沿a轴方向晶体结构,(c) Na1和Na2位点的配位环境30,(d)四方相Na3SbS4沿a轴晶体结构33Fig. 3 (a) The crystal structure of t-Na3SbS4 viewed alongc axis and (b) a axis, and (c) Na1 and Na2 coordination environments 30, (d) the crystal structure of c-Na3SbS4 viewed along a axis 33.
立方相Na3SbS4(c-Na3SbS4)晶胞中,a=b=c=0.71910 nm,Sb原子占据2a位点,S原子占据8e位点,Na原子占据6b位点,如图3d,SbS4四面体间隙组成三维钠离子传导通道33。Na3SbS4·H2O加热至150 °C便可脱去结晶水制得纯相c-Na3SbS4,冷却至室温,c-Na3SbS4可向t-Na3SbS4转变34,35。c-Na3SbS4室温离子电导率为2.8 mS·cm-1,比t-Na3SbS4稍高。研究表明,同c-Na3PS4,c-Na3SbS4也是高温稳定相。通过高能球磨制备的c-Na3SbS4冷却至室温后,c-Na3SbS4仍然是主相,并非t-Na3PS4,因此说明c-Na3SbS4也是室温稳定相,这可能与c-Na3SbS4的形成机理及微结构有关36。
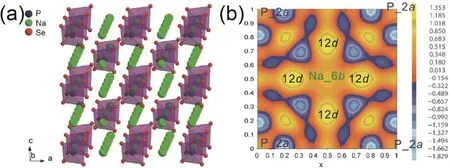
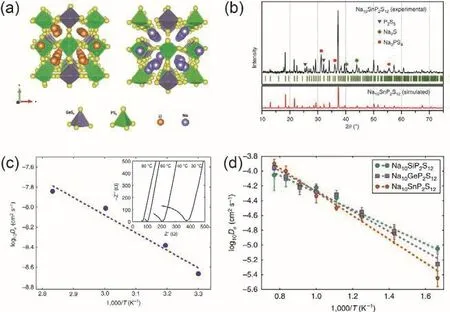
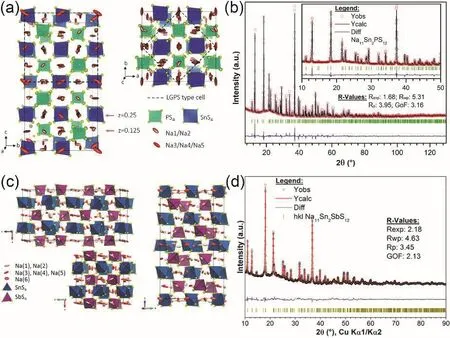
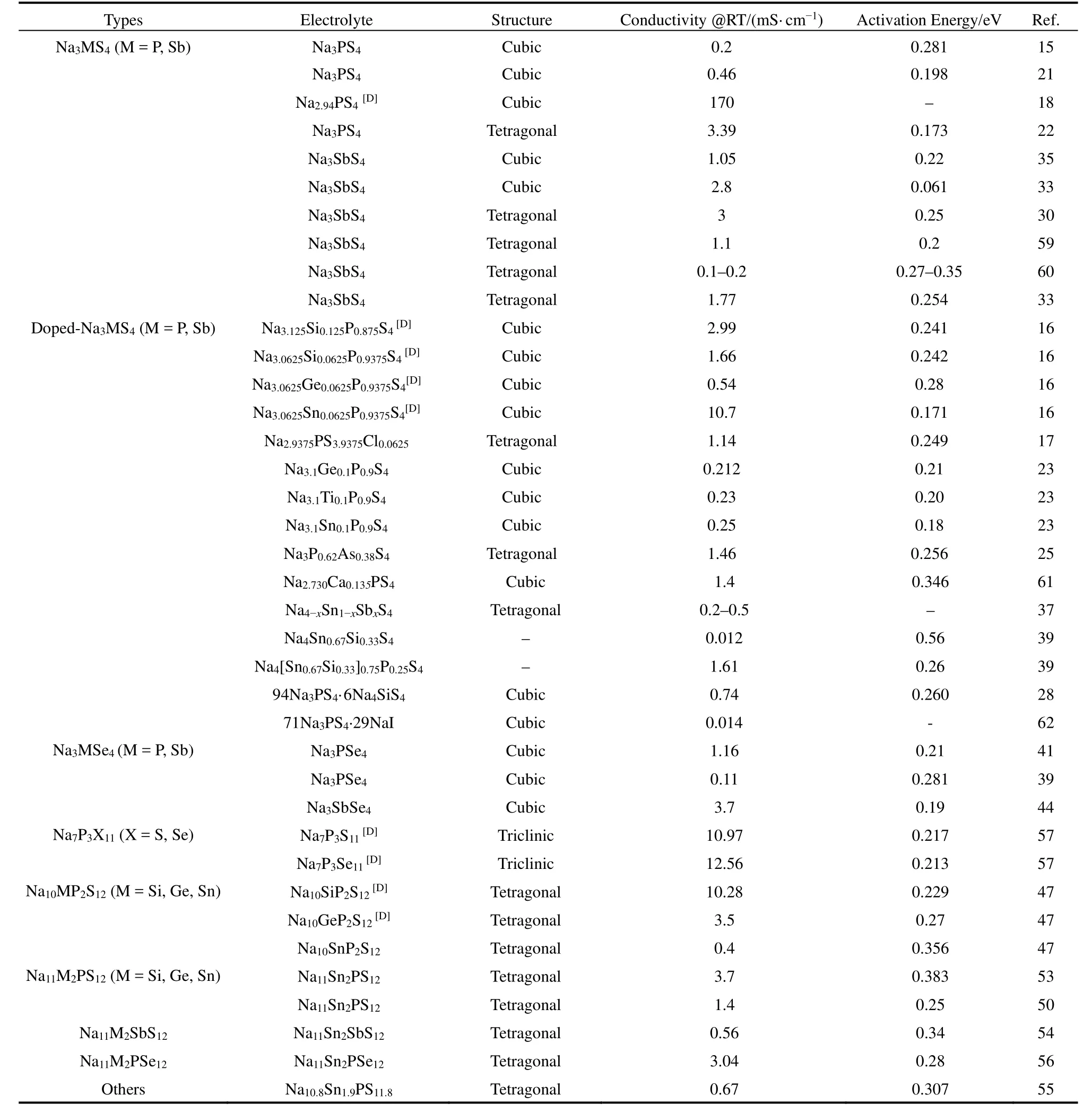
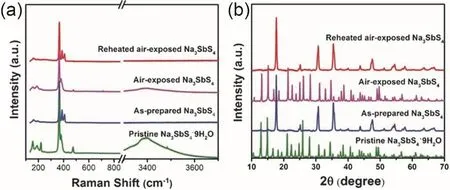
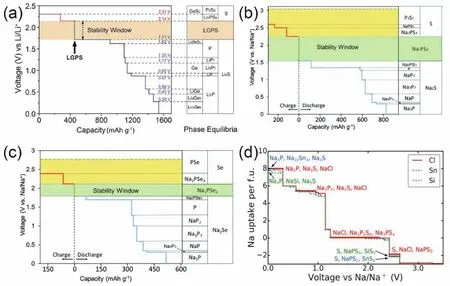
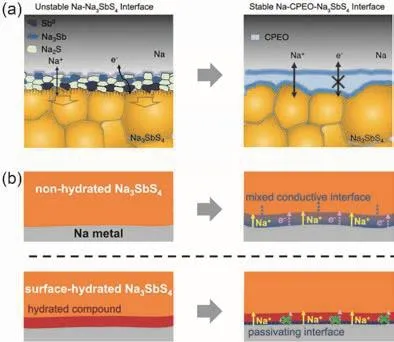
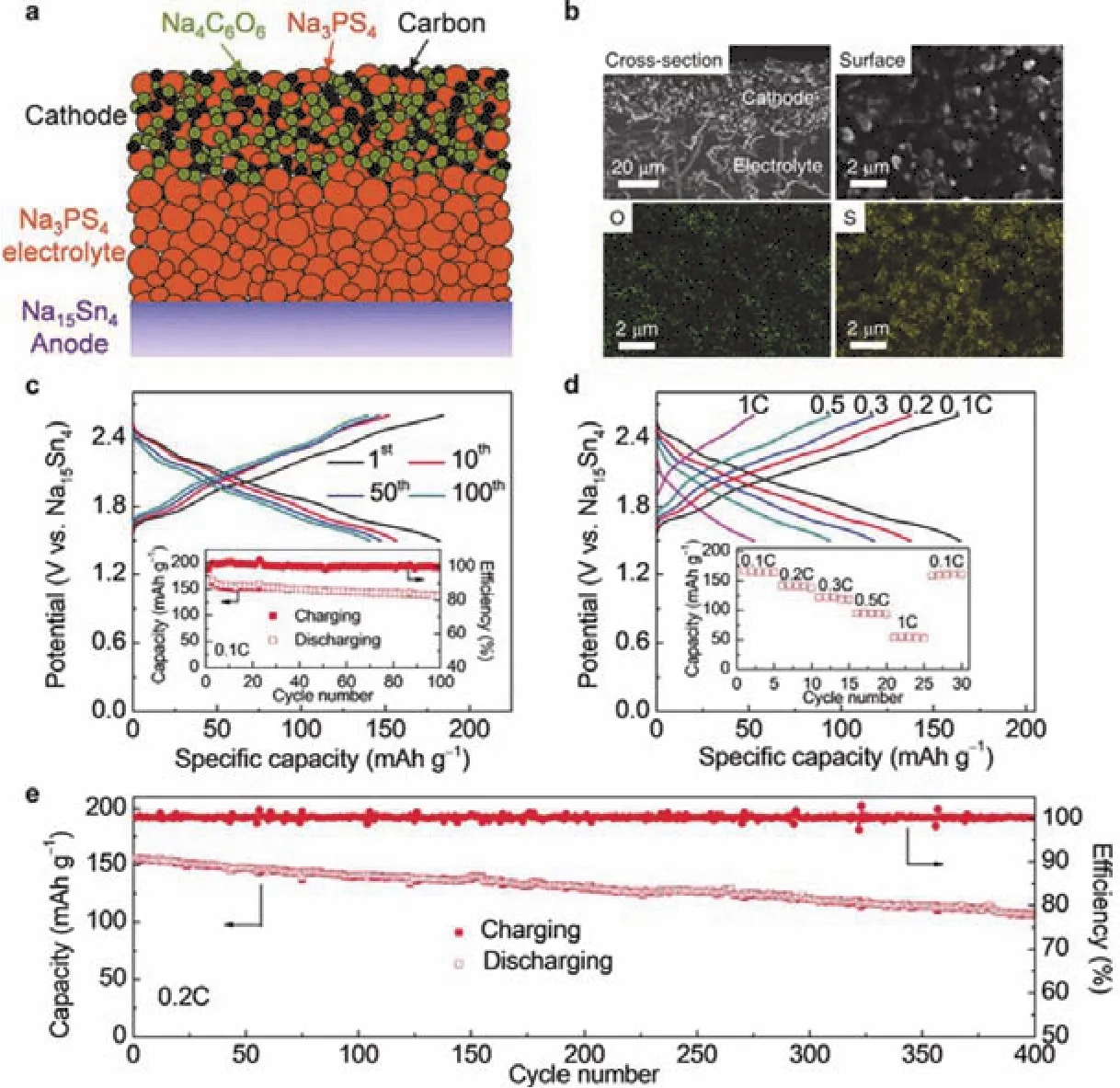
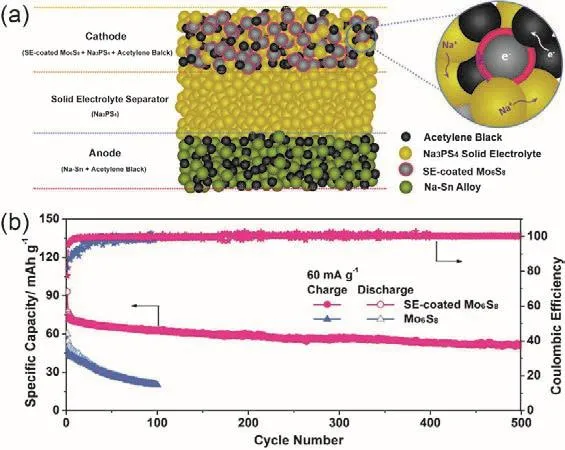
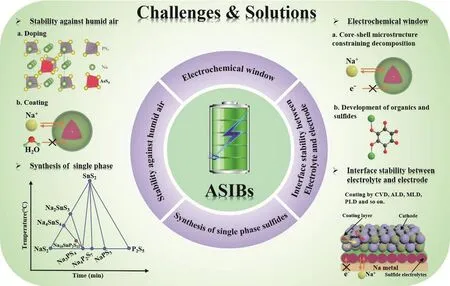
Sn取代Sb能够获得Na4-xSn1-xSbxS4(0.02 Tatsumisgo等29发现合适尺寸的扩散通道对于离子扩散是十分重要的,阴离子替换(S位)比阳离子(P位)替换对离子电导率的影响更显著。而且Se取代硫化物固体电解质中的S能提高电导率40,Se原子半径比S原子大,能够使晶格膨胀,离子扩散通道更适合快速传输;Se2-极化能力强,能削弱钠离子和阴离子之间的结合力。从这两个角度分析,Na3PSe4也应该是一种硫化物快离子导体41。 立方相Na3PSe4与立方相Na3PS4同构,晶胞参数比Na3PS4大4%。如图4,在立方相Na3PSe4晶胞中,P原子占据2a位点,Se原子占据8c位点,6b位点的Na+在PSe4四面体形成的扩散通道内跃迁41。与Na3PS4相似,Na+最有可能占据12d位点形成3维传输通道,其独特的电子密度分布提供快速的Na+输运路径21,41。立方相Na3PX4(X = S、Se)中的PX43-采取面心立方排布,钠离子占据X2-离子形成的四面体空穴,这种面心立方结构本征离子迁移数高32。但是在被活化的四面体位置的钠离子距离邻近钠离子仅有0.18 nm的距离,小于钠离子离子半径(~0.20 nm),不利于钠离子跃迁至瞬态位点。因此,同Na3PS4一样,完美的Na3PSe4也不是钠离子导体16。分子动力学模拟表明,Na+缺陷是立方相Na3PSe4能传导钠离子的关键,包括钠空位、钠间隙、弗伦克尔缺陷,其中钠空位缺陷的形成能最低,是钠离子传导的主要机制42。 图4 (a) Na3PSe4的晶体结构,PSe4四面体构成钠离子扩散通道,(b) xy平面(z = 0)电子密度分布傅里叶变换差分图41Fig. 4 (a) Crystal structure of Na3PSe4 illustrating the sodium ion at the diffusion channels formed by PSe4 tetrahedra.(b) Fourier map of the difference pattern of the xy-plane at z = 0 41. Na3PSe4与Na3PS4结构相似,因此,两者能够形成固溶体Na3PSxSe4-x。随着Se含量的增加,四方相Na3PSxSe4-x向立方相转变且晶体体积逐渐增大,Na—S/Se和P—S/Se键键长逐渐增大,离子电导率逐渐升高,活化能逐渐降低(图5)43。分子动力学模拟表明,与Na3PS4一样,钠离子在组成为化学计量比的Na3PSe4内是无法移动的,当引入2.1%钠空位后,不论是四方相还是立方相内钠离子的传输变得很容易。因此,Na3PSxSe4-x相结构不是影响钠离子的传导决定因素,钠空位和钠间隙等缺陷才是钠离子传导的关键。Na3PSxSe4-x组成的变化能引入空位,提高Na3PSe4的离子电导率。 Sb对P位进行取代得到单一立方相Na3P1-xSbxSe4,晶格参数随Sb含量的增加呈线性增长(图6a),Na3SbSe4晶格参数最大(a= 0.7492 nm)44。如图6b,Na3SbSe4晶胞中,Sb原子在2a位点,Se原子在8e位点,Na原子占据6b位点,两个SbSe4四面体形成正交的钠离子扩散通道。Na3SbSe4内钠离子的扩散通道比Na3PSe4的大,Na3PSe4内钠离子的扩散通道比Na3PS4大。因此,当立方相Na3P1-xSbxSe的P元素完全被Sb取代后离子电导率最高,活化能最低,说明更大的晶格参数更有利于钠离子的传导(图6c)。 2.3.1 Na10MP2S12 (M = Si, Ge, Sn) 图5 (a)晶胞体积和(b)键长与Se含量的变化关系,(c) Na3PSxSe4-x (x = 0, 1, 2, 3, 4)的电导率与温度之间的关系,(d) Na+扩散活化能与Se含量的变化关系43Fig. 5 (a) Cell volume (top) and (b) selected bond lengths versus Se concentration. The yellow and green shaded regions as shown in the top panel indicate the compositional ranges where Na3PSxSe4-x crystallizes in the tetragonal and cubic structures, respectively, (c) the conductivity (S cm-1) vs temperature plots for Na3PSxSe4-x (x = 0, 1, 2, 3, 4) are displayed on the top panel, (d) the activation energy for Na+ diffusion as a function of Se concentration 43. 图6 (a)晶格参数与Sb浓度之间的关系,(b) Na+在不同硫系化合物固态电解质中的扩散示意图,(c) Na3P1-xSbxSe4的电导率与温度之间的关系,(d) 室温离子电导率和活化能与Sb浓度之间的关系44Fig. 6 (a) Lattice parameter as a function of Sb concentration, (b) schematic diagram of Na ion diffusion in different compounds, (c) Arrhenius conductivity plots of Na3P1-xSbxSe4 in the temperature range of room temperature to 90 °C,(d) room temperature ionic conductivity (r) and activation energy (Ea) as a function of Sb concentration.The inset is Nyquist impedance plots of Na3P1-xSbxSe4 44. 图7 (a) Li10GeP2S12和Na10GeP2S12的晶体结构46,(b) Na10SnP2S12的X射线衍射实验图谱和模拟图谱,(c)电导率和温度之间的关系(插图为电化学阻抗谱),(d)分子动力学模拟计算Na10SiP2S12、Na10GeP2S12和Na10SnP2S12的电导率47Fig. 7 (a) Comparison of structures (unit cells) of Li10GeP2S12and the predicted Na10GeP2S12 compound 46.(b) experimental and simulated XRD patterns of Na10SnP2S12, (c) diffusivity calculated from experimentally measured ionic conductivity versus temperature. Dashed line is an Arrhenius fit to the data. (inset) Electrochemical impedance spectroscopy measurements, (d) Na-diffusivity in Na10SiP2S12, Na10GeP2S12 and Na10SnP2S12 from AIMD simulation.47. 如图7a所示,Na10GeP2S12(NGPS)与Li10GeP2S12(LGPS)晶体结构相似,由NaS6八面体,NaS4、PS4和(Ge0.5P0.5)S4四面体组成,其中,Na+沿着c轴一维隧道快速扩散。LGPS具有超高的室温离子电导率(12 mS·cm-1),能与液态电解质相匹敌13,与LGPS同构的Li9.54Si1.74P1.44S11.7Cl0.3室温离子电导率甚至高达25 mS·cm-1,是目前报道的室温离子电导率最高的固体电解质45。量子密度泛函理论计算得出NGPS同样具有超高的室温离子电导率(4.7 mS·cm-1)46,然而目前想要成功合成出高电导率的纯相Na10MP2S12还是比较困难的,具体原因还不是很清楚。Richards等47用固相法制得了四方相Na10SnP2S12,快速冷却能得到纯相的Na10SnP2S12,电导率低;慢速冷却时虽产生少量Na2S、P2S5、Na3PS4杂相,但室温离子电导率能达0.4 mS·cm-1,与c-Na3PS4相当(图7b,c)。在慢速冷却产物Na10SnP2S12中有更高的Sn/P含量比,导致晶体有更大的体积,因此获得了更高的室温离子电导率,LGPS电解质中也存在这种由晶体体积不同引起离子电导率产生较大差异的现象。在LMPS和NMPS电解质中(M = Si, Ge, Sn),Si基电解质活化能比Ge基低,Ge基电解质活化能比Sn基低,因此Na10SiP2S12可能有更高的室温离子电导率(图7d)。Kandagal等48通过理论计算的得出Na10GeP2S12室温离子电导率为4.7 mS·cm-1,而Hayashi等49通过固相球磨并在235 °C下进行热处理制得的Na10GeP2S12室温离子电导率为0.024 mS·cm-1,比Richards理论计算得到的值低一个数量级,比Kandagal计算得出的4.7 mS·cm-1低两个数量级,作者认为可能的原因有两方面:(1)低温相Na10GeP2S12经过有序-无序转化后离子电导率可能会有所提高;(2) 600 K温度下由分子动力学模拟得到的离子电导率值与室温测得的实验值之间的会存在差异。 2.3.2 Na11M2PS12 (M = Si, Ge, Sn) Nazar等50对Na-Sn-P-S的相图进行研究发现,在前驱体Na2S、P2S5、SnS2熔融后以缓慢速率冷却可以得到无色矩形单晶。单晶衍射解析出产物的晶体结构为Na11Sn2PS12新相。Na11Sn2PS12室温离子电导率1.4 mS·cm-1,活化能0.25 eV,比Na10SnP2S12低0.1 eV。如图8a所示,晶体结构与Li10GeP2S12(LGPS)相似13,51,52,SnS4、PS4四面体构成沿c轴和ab平面的三维钠离子传导通道,但不同的是,Na+只占据五种八面体位点,含有部分空位的Na1、Na2位点与几乎完全占据的Na3、Na4、Na5位点交叉,NaS6八面体通过共面形成四通八达的传输路径,使得钠离子能够快速传导。多晶衍射(图8b)明确产物是Na11Sn2PS12纯相,然而用同样的合成方法、相同的前驱体以特定的化学计量比制得的Na10SnP2S12含有40% Na3PS4杂相,说明热力学上 Na11Sn2PS12比Na10SnP2S12稳定。 同一时期,Roling等53发现前驱体Na3PS4和Na2SnS4中Sn : P摩尔比为1 : 2时无法得到Na10SnP2S12纯相,提高Sn : P摩尔比能降低杂相Na3PS4的含量,当摩尔比等于2 : 1时,正好得到Na11Sn2PS12纯相,室温离子电导率高达4 mS·cm-1。同步辐射测试证明了存在钠空位,且价键理论证实空位连接成的三维传输通道使得钠离子能够快速传导,因此具有与LGPS几乎同等优异的离子传导性能。 图8 (a) Na11Sn2PS12的晶体结构,(b)纯相Na11Sn2PS12多晶粉末衍射及精修50;(c) Na11Sn2SbS12的晶体结构,(d)纯相Na11Sn2SbS12多晶粉末衍射及精修54Fig. 8 (a) The framework of Na11Sn2PS12 and (b) Rietveld refinement of X-ray powder diffraction data of single phase polycrystalline Na11Sn2PS12 50. (c) The framework of Na11Sn2PS12 Na11Sn2SbS12 and (d) Rietveld refinement of X-ray powder diffraction data of single phase polycrystalline Na11Sn2SbS12 54. 用等价的Sb5+取代Na11Sn2PS12中P5+可得到一种新的钠离子硫系化合物固态电解质Na11Sn2SbS1254。四方相Na11Sn2SbS12晶体结构和粉末衍射图谱如图8c,d所示,钠离子分布在6种位点上,其中5种位点上的钠离子与S形成不规则的NaS6八面体,Na—S键键长为0.32–0.35 nm不等,剩下的钠占据间隙位点Na(6),钠离子在Na11Sn2SbS12晶格内等间距输运。测得室温离子电导率为0.56 mS·cm-1,不及Na11Sn2PS12,可能是由于P—S和Sb—S局部键和效应不同对钠离子传输的影响不同所致。密度泛函理论计算可知Sb替换P后,Sb—S键键长更长,键能更低,Sb5+电负性(1.8)比P5+电负性(2.1)更低,因此,S2-电子密度更高,Na+与S2-之间库伦吸引作用更强,使Na+结合更紧密。这种诱导效应使得钠离子移动性变差,离子电导率变差。然而,Na11Sn2SbS12的空气稳定性优于Na11Sn2PS12。暴露在干燥的空气中36 h后,XRD衍射表征显示Na11Sn2PS12已降解,而Na11Sn2SbS12没有明显变化,离子电导率稍有降低。 Wang等55发现在Na10SnP2S12合成过程中,随着Sn/P摩尔比增大,Na3PS4杂质相的比例逐渐减小,当Sn/P等于1.9时,得到热力学和动力学均稳定的纯相Na10.8Sn1.9PS11.8。与Na11Sn2PS12结构一样,SnS4、PS4四面体构成沿a轴、b轴和c轴的三维钠离子传导通道,空位传导为主导的Na10.8Sn1.9PS11.8室温离子电导率0.67 mS·cm-1,介于Na10SnP2S12和Na11Sn2PS12之间,活化能0.307 eV。 表1 钠离子硫系化合物固态电解质离子传导性能Table 1 Summary of chalcogenide electrolytes for all-solid-state sodium ion batteries. 2.3.3 Na11M2PSe12 (M = Si, Ge, Sn) Na11Sn2PSe12与Na11Sn2PS12同构,除了占据着由4个Se2-离子和2个Na+形成的Na(6)位点外,其他所有的钠离子占据着由6个Se2-离子形成的八面体位点,NaSe6八面体之间通过共面连接形成3D传输网络56。Na11Sn2PSe12室温离子电导率2.15 mS·cm-1,作者认为3D传输网络、高Na+空位率(高达16%)和Na—Se键之间的弱相互作用是Na11Sn2PSe12具有高离子电导率的主要原因。 受Li7P3X11(X = S、Se)的启发,Ceder等57认为钠离子也可以在面心立方阴离子框架结构的邻近四面体位点间直接迁移,因此,Na7P3X11(X = S、Se)也应有高离子电导率。分子动力学模拟结果表明,Na7P3S11和Na7P3Se11室温离子电导率高高于10 mS·cm-1。然而,氧的可极化性比硫和硒弱,Na+和P5+离子间的距离比在Na7P3S11和Na7P3Se11更短,因此,阳离子间更强的作用力导致在Na7P3O11能量谱中Na的位点能变化大,Na+迁移需要的活化能更高,因此Na7P3O11的室温离子电导率仅为0.003 mS·cm-1。 综上,目前钠离子硫系化合物固态电解质主要分为以Na3PS4为代表的硫系化合物固态电解质Na3MX4(M = P、Sb;X = S、Se)、硫系化合物固态电解质NamMxPyX12(M = Si,Ge,Sn;X = S,Se;10 ≤m≤ 11;x/y= 1/2或2)和Na7P3X11(X = S,Se)四种类型。整体来看,多数硫系化合物固态电解质室温离子电导率高于10-4S·cm-1,基本能满足固态电池的要求。也有的硫系化合物电解质甚至高于10-3S·cm-1,能与液态电解质相匹敌,表1对现有报导的硫系化合物固态电解质离子传导性能进行了总结。为避免自放电和正负极短路,通常要求硫系化合物电解质的电子电导率要比离子电导率低几个数量级,此外,为抑制金属负极枝晶的形成,在1和10 mA·cm-1沉积电流密度下要求电子电导率分别低于10-10和10-12S·cm-158。 离子电导率只是影响全固态电池发展的重要因素之一,硫系化合物固态电解质的稳定性、与电极兼容性等问题均是硫系化合物固态电解质基全固态钠离子电池发展所面临的挑战,包括了硫化物本征化学稳定性、电化学稳定性、电极/电解质界面稳定性等。 硫系化合物固态电解质在空气中易被潮解或氧化,容易生成有毒的H2S气体,电导率会急剧下降33,63,64。另外,硫化物的稳定性遵循软硬酸碱理论:硬酸优先与硬碱反应,软酸优先与软碱反应。与硬酸相比,软酸原子半径更大,电子分布更分散,更容易极化。具体地,对于硫代磷酸盐电解质(Na3PS4),氧属于硬碱,优先与磷反应(硬酸)替换软碱(S),因此硫代磷酸盐在空气中不稳定64。 图9 Na3SbS4·9H2O、Na3SbS4、在空气中暴露48 h后的Na3SbS4、暴露于空气后再进行150 °C真空干燥1 h后的Na3SbS4样品的(a)拉曼光谱和(b) X射线衍射图谱35。Fig. 9 (a) Raman spectra, (b) XRD patterns of pristine Na3SbS4·9H2O, as-synthesized Na3SbS4, air-exposed Na3SbS4 (48 h), and reheated air exposed Na3SbS4 sample (150 °C for 1 h under vacuum) 35. 基于软硬酸碱理论,Sb与P相比是软酸,与氧(硬碱)的亲和力降低,在干燥的空气中Na3SbS4比Na3PS4更稳定。Na3SbS4对CO2也是稳定的,在CO2气流中存放24 h,XRD衍射图谱基本没有变化且电导率几乎没有衰减。但是,Na3SbS4在潮湿的空气中容易形成水合物(Na3SbS4·9H2O),经过150–200 °C真空热处理能够重新变成Na3SbS4,如图9拉曼光谱和XRD图谱所示,电导率几乎没有衰减35。因此,除了固相法合成Na3SbS4外,也能用液相法制备Na3SbS4,利于界面工程的实施59,60。据报道,合金元素As部分取代P也能增强Na3PS4的氧气和湿度稳定性25,65。Na3P1-xAsxS4在140 °C、湿度15%的空气中暴露100 h后XRD图谱没有明显变化,对湿度的稳定性也随As含量的升高而增强。与P—O相比,As—O之间亲和力较弱,As部分取代后Na3P1-xAsxS4与水的反应产物不再是容易生成的Na3POS3、Na3PO2S2和H2S,而是难生成的Na3P1-xAsxS4·nH2O (n= 8或9)65。 电化学窗口是电解质又一重要性质,电化学窗口足够宽时电极材料才有更多的选择,足够高时高压正极材料才能输出高比能。线性极化法测Na/硫系化合物固态电解质/惰性金属电极组成的半阻塞电极的电化学稳定窗口为0–5 V15,25,35,62,即使离子电导率与液态电解质相当,然而固态电池性能远不如液态电池。这是因为在这种半阻塞电极内,电解质与惰性电极(集流体)接触面小、电解质电子电导很低,导致电解质的分解反应动力学速率很慢;然而在复合电极中,电极材料、导电炭黑和电解质混合在一起,电解质与电极接触且电子电导得到改善,电解质的氧化还原速率缓慢加速,造成循环稳定性下降,因此这种方法常常高估了电解质的电化学稳定窗口65,67,当电解质与惰性电极紧密接触时,比如,电解质溅射到惰性电极表面一侧的情况,用这种线性极化法测得的电解质窗口才比较准确。传统的半阻塞电极测得的锂硫代磷酸盐电化学窗口为0–5 V,但是理论计算得出的窗口只有1.7–2.1 V (图10a),因为P5+有很高的还原电势,S2-有很低的氧化电势68。在钠硫代磷酸盐电解质中也存在高估电解质窗口的现象。第一性原理计算表明 Na3PS4的电化学稳定窗口为1.55–2.25 V67。如图10b所示,1.55 V以下,Na3PS4被还原成 Na2S和 Na—P化合物;2.25 V以上,Na3PS4分解生成 P2S7、S等缺钠相。如图 10c所示,Na3PSe4的电化学稳定窗口为1.80–2.15 V,1.80 V以下,分解生成Na2Se和Na—P化合物;2.15 V以上生成化合物PSe和单质Se67。在电压高于2.4 V时,经掺杂改性的Na3PS4(Na2.9375PS3.9375Cl0.0625)不稳定,分解生成NaPS3、S和NaCl (图10d)。将Na3PS4、Na3PSe4与导电碳混合作电极进行充放电实验,得出的电化学稳定区间与理论计算基本吻合,进一步验证了半阻塞电极的不准确性。 图10 第一性原理计算得出的电化学稳定窗口及相平衡产物:(a) LGPS 68,(b) Na3PS4 67,(c)Na3PSe4 67,(d) Cl、Sn、Si掺杂的Na3PS4 17Fig. 10 The first principles calculation results of the voltage profile and phase equilibria products of (a) LGPS 68(b) Na3PS4 67, (c) Na3PSe4 67 and (d) the doped Na3PS4 compounds by Cl, Sn, Si 17. 图11 全固态钠离子电池界面兼容和稳定性问题及实验表征方法67Fig. 11 Summary of compatibility and stability problems in all-solid-state sodium batteries and the experimental methods for assessing these issues 67. 尽管硫系化合物电解质的离子电导率取得了显著的进展,但是全固态电池的界面内阻很高,特别是采用高压氧化物正极和碱金属负极的全固态电池,循环稳定性和倍率性能很差63,68。化学不兼容、电化学反应、机械接触不好都能增加界面内阻,由化学或电化学引起的界面不稳定能改变电荷转移动力学、增加过电势,降低固态电池的循环稳定性和倍率性能。如图11所示,Ceder等67总结了硫系化合物固态电解质Na3PX4(X = S,Se)的电化学稳定性、与正负极的界面稳定性及实验表征方法,为今后的研究提供了思路。 3.3.1 硫系化合物固态电解质与正极的界面稳定性 正极与电解质之间绝对的稳定是很困难的,正极和电解质反应的产物在正极/电解质界面形成有害或有利的界面层,有利的界面层必须是自钝化的且允许钠离子快速穿过。计算表明,Na3PS4、Na3PSe4与层状氧化物正极NaxMnO2(M = Cr、Mn、Fe、Co、Ni)可能的反应分两种:(1)阴离子交换反应生成NaCrX2(X = S、Se)和Na3PO4,(2)复杂的氧化还原反应生成三种以上分解产物67。差示扫描量热与热重联用技术和XRD实验结果表明NaCrO2与Na3PS4、Na3PSe4室温下相对稳定67,因此有硫化物固态钠离子电池研究也可用NaCrO2作正极21,22,33,58。 图12 (a)循环后Na3PS4、Na2.9375PS3.9375Cl0.0625、Na2.875PS3.875Cl0.125电解质表面S 2p, P 2p, Cl 2p的X射线光电子能谱,(b–d) Na3PS4、Na2.9375PS3.9375Cl0.0625、Na2.875PS3.875Cl0.125电解质在Na金属对电池中的循环稳定性84Fig. 12 (a) X-ray photoelectron spectrum of solid electrolyte interface, S 2p, P 2p, and Cl 2p region of after cycling of Na3PS4, Na2.9375PS3.9375Cl0.0625, Na2.875PS3.875Cl0.125, (b–d) galvanic square-wave cycling of Na metal symmetric cell using Na3PS4, Na2.9375PS3.9375Cl0.0625, Na2.875PS3.875Cl0.125 as solid state electrolyte, respectively 84. 3.3.2 硫系化合物固态电解质与金属钠负极的界面稳定性 钠金属作钠离子电池负极有最低的还原电势(-2.714 V)、最高的理论容量(1165 mAh·g-1),室温Na-S电池和Na-O2电池用金属钠作负极,比传统氧化物做正极、碳材料作负极的钠离子电池的能量密度高70–74。此外,与硬碳等传统负极材料相比75–77,用金属钠作负极,正极材料将会有更多的选择。因此,用金属钠作负极是未来发展高能量密度钠二次电池的重要方向78–80。固态金属钠二次电池中钠枝晶能得到有效抑制,安全性更高。然而,电解质和金属钠界面稳定性仍然是巨大的挑战,尤其是硫系化合物固态电解质与金属钠的界面稳定性。 Na3PS4与金属钠的界面很不稳定,与Na反应生成电子绝缘、高离子电导的Na2S和离子、电子混合传导的Na3P,因此,电子渗流持续发生,界面副反应持续发生,界面阻抗逐渐增大,电池比容量和循环寿命持续衰减81–83。同样,Na3SbS4与Na反应生成Na2S和Na3Sb混合离子-电子导电相,值得庆幸的是,除生成Na2S和Na3P,Na2.9375PS3.9375Cl0.0625与Na在界面还能生成NaCl (图12a),能够钝化、稳定界面,缓解Na3PS4分解,并且Cl掺杂量为6.25%时面电阻最小,过电位最小,Cl掺杂量过多(12.5%),也会增大面电阻,增大过电位(图12b–d)84。 混合离子-电子传导界面中间相允许电子通过电解质输运,导致电池自放电,严重影响固态电池的电化学性能85。如果可以构建电子阻挡层,切断硫系化合物固态电解质分解反应的电子渗流通道,就可以稳定电解质/金属Na界面。目前,有两种方法构建电子阻挡层。如图13a所示,Yao等86在Na3SbS4与金属Na中间引入纤维素/PEO混合聚合物中间层,有效阻挡了电子渗流,Na3SbS4不再分解,界面稳定,60 °C下金属Na能以0.1 mA·cm-2稳定沉积剥落800次。Ceder等87发现Na3SbS4在空气中吸潮表面能生成Na3SbS4·8H2O水合物,Na3SbS4表面的Na3SbS4·8H2O与金属Na反应生成钝化产物NaH和Na2O,减少了具有电子导电能力的Na3Sb相,阻止电解质Na3SbS4连续分解(图13b)。 图13 (a)不稳定的Na-Na3SbS4界面和稳定的Na-CPEO-Na3SbS4界面86,(b)固体电解质-钠金属界面电化学循环前(左)和后(右)示意图。非水合Na3SbS4 (顶部)在循环过程中形成了混合导电界面层,而水合Na3SbS4 (底部)在循环过程中形成了钝化界面87Fig. 13 (a) Schematic illustration of unstable Na-Na3SbS4 interface and stable Na-CPEO-Na3SbS4 interface 86.(b) schematic illustration of solid electrolyte-Na metal interface before (left) and after (right) electrochemical cycling. A mixed conductive interface layer grew upon cycling of the non-hydrated Na3SbS4 (top), whereas a passivating interface was formed on the hydrated compound of the surface-hydrated Na3SbS4 (bottom) 87. 除了上述提到的硫系化合物固态电解质与多数氧化物正极材料不稳定外,硫系化合物固态电解质的电化学窗口窄,分解电压低于NaFeO488、Na3V2(PO4)389–93、Na2/3Co2/3Mn1/3O294等大多数嵌入型正极材料的工作电压,因此,硫系化合物固态电解质更适合于工作电位低的正极材料,比如TiS215,17,25,37,62,95、FeS259,96、S97,98、Na2S99等转化型正极材料。但这些转化型正极材料体积膨胀大,循环稳定性不佳。与金属Na负极相比,Na-Sn合金与硫系化合物固态电解质的界面兼容性更好,是目前多数硫化物固态钠离子电池选用的负极材料16,21,25,33,37,49–51,53,55。Yao等100用有机嵌入型Na4C6O6既作正极,Na15Sn4合金作负极,与Na3PS4有良好的化学、电化学稳定性。如图14所示,全固态钠离子电池在60 °C下工作具有184 mAh·g-1比容量,0.1C倍率下循环100周容量保持率为76%,0.2C倍率下循环400周容量保持率仍有70%,具有良好的循环稳定性。 图14 (a)Na4C6O6|Na3PS4|Na15Sn4全固态钠离子电池示意图,(b)电极/电解质界面、正极表面SEM图片及O和S的EDX图片,(c) 60 °C下,0.1C倍率不同周次的充放电曲线(插图为0.1C电流密度下的循环性能),(d)不同倍率下(0.1C–1C)的充放电曲线及循环性能,(e) 60 °C下,0.2C长循环比容量及库伦效率100Fig. 14 (a) Schematic of the Na4C6O6|Na3PS4|Na15Sn4 ASIBs, (b) SEM image of cathode/electrolyte cross-section and cathode surface and corresponding EDX mapping of O and S (bottom); (c) Charge/discharge voltage profiles at different cycle numbers at 0.1C at 60 °C (Inset: Capacity and coulombic efficiency vs cycle number at 0.1C); (d) Representative charge/discharge voltage profiles at different current rates (Inset: Rate capabilities and cycling of the battery from 0.1C to 1C); (e) Capacity and coulombic efficiency vs cycle number at 0.2C at 60 °C 100. 为确保全固态电池能稳定循环,在解决电极/电解质电化学不稳定的同时,还需要考虑电极/电解质界面接触不充分、充放电过程中Li+/Na+嵌入/脱出引起的应力/应变问题。硫化物玻璃/玻璃-陶瓷电解质具有良好的冷静压成型性能,与电极界面接触紧密,电极/电解质界面阻抗小。电池充放电过程中,Li+/Na+嵌入/脱出伴随电极活性材料体积膨胀收缩,容易导致颗粒破裂形成裂纹,晶界阻抗逐步增大,从而导致电池容量衰减。硫系化合物固态电解质的杨氏模量介于氧化物陶瓷电解质和聚合物电解质之间,具有良好弹性的变形能力,在电极片内混入一定量的硫系化合物固态电解质不仅能填充正极内部颗粒间间隙,还能够缓冲电极材料的体积膨胀,因此多数硫系化合物固态电解质的固态电池具有良好的循环性能95,101–103。与氧化物陶瓷电解质混合冷静压可以填充Na3Zr2Si2PO12陶瓷内部孔隙,混合电解质片的致密度高达80%,钠离子输运连续顺畅,界面阻抗仅15.8 Ω·cm-1104。另外,体积变化产生的应力主要集中在电极/电解质固/固界面上。良好的弹性变形能力能缓冲固固界面处的应力,保持电解质和电极界面的良好接触。 制作全固态电极/电解质混合电极要求电极内部电极材料和电解质充分接触才能减小内部阻抗。有研究表明,手动研磨混合无法使S和Na3PS4混合均匀,电极内部仍然存在间隙;电极材料S、导电剂AB、电解质Na3PS4经球磨混合后各组分均匀分布,室温固态Na-S电池表现出比高温固态Na-S电池更好的电化学性能,如图15a,b所示97。在S电极内混入非晶P2S5,电化学原位反应生成的Na3PS4与S电极混合更均匀,全固态电池性能比S和Na3PS4直接混合的全固态电池性能更好,如图15c,d所示98。 另外,硫系化合物固态电解质可用液相法制备,在电极材料颗粒表面包覆一层硫系化合物固态电解质,原来电极材料颗粒与电解质颗粒之间的点接触变成面接触,全固态电池性能更佳。如图16所示,液相法制备的Na3PS4包覆在Mo6S8正极颗粒表面,更充分的面接触增加了Na+和电子传导,增加了电荷转移反应活性位点,活性材料利用率更 图15 (a,b)手磨混合的复合正极(S + AB + Na3PS4)及球磨混合的复合正极(S-AB-Na3PS4)的SEM图及元素分布97,(c,d) S与Na3PS4混合的复合S-KB-Na3PS4正极,与P2S5混合的S-KB-P2S5复合正极放电后的SEM图及元素分布98Fig. 15 SEM and EDX mapping images of the sulfur composite electrodes prepared by hand mixing[(a) S + AB + Na3PS4] or mechanical milling [(b) S-AB-Na3PS4] 97; (c) a surface of the S-KB-Na3PS4 and(d) S-KB-P2S5 composite electrodes after the discharge process 98. 图16 (a) Na3PS4包覆的Mo6S8正极及块状全固态钠离子电池示意图,(b) 60 °C下,经Na3PS4包覆的Mo6S8和没有包覆的Mo6S8在全固态钠离子电池中的循环性能105Fig. 16 (a) Schematic diagram of a bulk-type all solid state sodium ion battery enabled by a Na3PS4-coated Mo6S8 cathode, (b) cycling performances and Coulombic efficiencies of the Mo6S8 and Na3PS4-coated Mo6S8 cathodes in all solid state sodium ion battery at 60 °C 105. 本文综述了钠离子硫系化合物固态电解质晶体结构、电导率、稳定性及在全固态钠离子电池应用方面的进展,典型的钠离子硫系化合物固态电解质的电导率、空气稳定性、界面稳定性、电化学窗口及制备难易程度五个方面的性质对比如图17所示。 总的来说,由于硫族阴离子的强极化特性,硫化物比氧化物具有更高的室温离子电导率;硫化物玻璃/玻璃-陶瓷具有优越的力学性能,冷压易成型,固体颗粒在晶界和电极/电解质界面接触更充高,比容量更高,且在60 °C下能稳定循环500周,是目前循环性能最好的全固态钠离子电池105。分,离子传导顺畅,是一类很有发展前景的固态电解质。然而,硫系化合物固态电解质的发展仍然面临一些挑战,我们总结如下(图18): 图17 典型硫系化合物固态电解质在电导率、空气稳定性、界面稳定性、电化学窗口及制备难易程度等方面的对比Fig. 17 Comparison of typical chalcogenide electrolytes for ASIBs on conductivity, stability against humid air, interface stability between electrode and electrolyte, electrochemical window and preparation technology. 图18 钠离子硫系化合物固态电解质发展面临的挑战及解决方案Fig. 18 Challenges and solutions for development of chalcogenide electrolytes s for ASIBs. (1)对空气敏感,在湿度高的环境中容易潮解释放有毒的H2S气体、在干燥空气中容易被氧化,因此硫系化合物固态电解质要求在惰性气氛下处理,增加了操作难度; (2)硫系化合物固态电解质中S或Se容易与氧化物正极的中O发生阴离子交换,高价P/Sb/Sn/Ge容易被金属Na还原,因此与氧化物正极材料界面氧化稳定性及金属钠负极界面还原稳定性仍需改善; (3)电化学窗口窄且分解电压低,很多常规高电压正极材料不适用;绝大多数固态电池用Na-Sn合金作负极,碳基负极材料难与其匹配; (4)计算表明Na10GeP2S12、Na7P3X11(X = O、S、Se)电解质的电导率高,但是实验合成纯相的Na10GeP2S12、Na7P3X11(X = O、S、Se)仍比较困难。 我们对于解决上述挑战的方案也进行了总结(图18): (1)基于软硬酸碱理论进行元素掺杂、表面包覆一层氧化物保护层或者与聚合物及进行复合提升空气稳定性; (2)采用固体核磁等先进原位表征技术对界面化学成分进行动态分析,利用化学气相沉积、原子层沉积、分子层沉积和磁控溅射等先进制备技术进行表面包覆调控; (3)设计壳-核结构限制硫化物体积膨胀时的分解106,开发有机物、硫化物电极材料; (4)结合相图,在熔融淬火过程中对降温速率进行精确控制;利用湿法化学进行液相合成并提纯。2.2 Na3MSe4 (M = P, Sb)系列
2.3 NMPX (M = Si, Ge, Sn; X = S, Se)系列


2.4 其他钠离子硫系化合物固态电解质
3 硫系化合物固态电解质的稳定性
3.1 硫系化合物固态电解质的本征化学稳定性
3.2 硫系化合物固态电解质的电化学稳定性
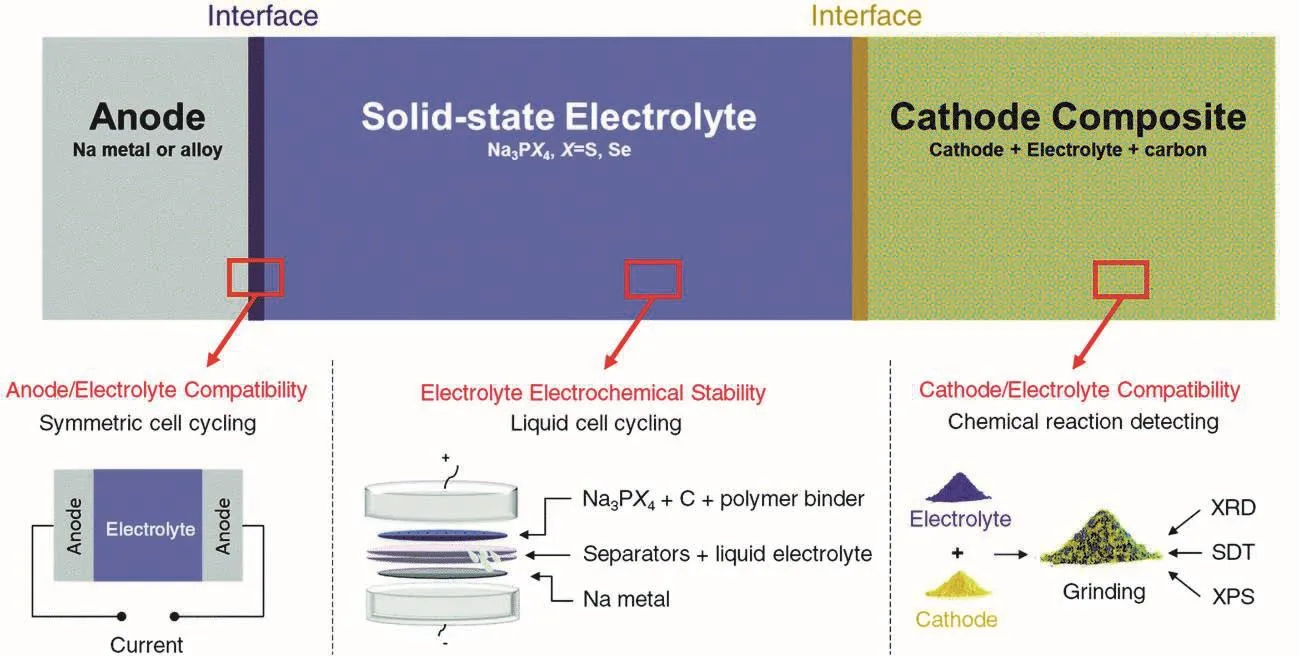
3.3 硫系化合物固态电解质与电极的界面稳定性

4 硫系化合物固态电解质基全固态钠离子电池

5 总结与展望

猜你喜欢
仪器仪表用户(2021年10期)2021-11-27 08:26:14
上海理工大学学报(2021年3期)2021-07-20 08:04:04
陶瓷学报(2021年1期)2021-04-13 01:33:40
陶瓷学报(2021年1期)2021-04-13 01:32:54
中学生数理化(高中版.高二数学)(2017年1期)2017-04-16 05:33:49
中华老年多器官疾病杂志(2016年2期)2016-01-16 03:15:46
电源技术(2015年2期)2015-08-22 11:28:30
电源技术(2015年9期)2015-06-05 09:36:06
导航定位学报(2015年2期)2015-06-05 09:27:42
物理化学学报(2015年5期)2015-02-28 17:34:57
