
MYBPC3基因突变致儿童孤立性心肌致密化不全1例并文献复习
2020-06-14 08:04周洋洋成举森李树军
河南医学研究 2020年14期
周洋洋,成举森,李树军
(1.郑州人民医院 儿科,河南 郑州 450000;2.新乡医学院第一附属医院 a.小儿内三科;b.PICU,河南 新乡 453100)
孤立性心肌致密化不全(isolated ventricular non-compac tion,IVNC)是一种特殊类型的心肌病,普遍认为心脏胚胎期心肌纤维致密化过程异常终止,导致两层(致密化层和非致密化层)心肌的形成。其表现为心室内异常粗大的肌小梁和交错的深隐窝为特征的一种心肌病,又称海绵样心肌、胚胎样心肌,主要累及左室,累及右室或者双室者少见。该病具有家族遗传倾向,在儿童患者中尤其明显。2008年欧洲心脏病学会(Europe an Society of Cardiology,ESC)发表声明,将其归类为原发性遗传性心肌病。由于临床表现不典型,极易误漏诊。现将收集的1例IVNC报道如下。
1 临床资料
患儿,女,5个月余,因“呼吸困难10 d,加重2 h”于2018年7月9日收住新乡医学院第一附属医院儿童重症监护室。2018年6月30日患儿无明显诱因(10 d前)出现呼吸困难,伴阵发性咳嗽、喘息,当地县医院以“支气管肺炎”处理,效差,外院查心脏超声疑似心肌致密化不全,2 h前呼吸困难加重,面色苍白,口周发绀,点头呼吸,转至新乡医学院第一附属医院PICU。既往史,家族史无特殊。其哥哥3岁,体健。体格检查:体温为37 7℃,心率为每分钟160次,呼吸频率为每分钟32次,体质量为6 6 kg,血压为 80/62 mm Hg,神志清,精神差,面色苍白,口唇发绀,点头呼吸,鼻翼煽动,鼻孔扩张,双肺呼吸音粗糙,可闻及喘鸣音及中细湿 音。心律齐,心音低顿,各瓣膜未闻及杂音,肝脾未触及肿大,神经系统查体未见明显阳性体征。查血常规、血肝肾功能、电解质、心肌酶谱均基本正常。血气分析提示:pH为 7 48,PaO2为 107 mm Hg,PaCO2为54 mm Hg,剩余碱为15 7 mmol·L-1,提示代谢性碱中毒。心脏超声示:射血分数为50%,左室大,余房腔大小及血管内径正常。左室心肌呈蜂窝样改变,左室后壁厚度正常,室壁运动减弱,肺动脉压力为40 mm Hg,提示左室致密化不全,左心功能低、肺动脉高压。期间完善基因诊断,提示:MYBPC3基因存在突变位点(c 2671C>T),属于错义突变,且其父亲与哥哥为杂合突变型,但均未发病(见图1)。入院后立即予持续气道正压通气辅助呼吸,预防性应用3 d头孢他啶注射液抗感染,雾化祛痰,精氨酸纠正代谢性碱中毒等治疗,并予西地兰强心及磷酸肌酸等营养心肌治疗后,2018年7月24日好转出院,院外长期口服地高辛口服液、卡托普利、潘生丁,建议长期随访心脏超声、地高辛药物浓度、心肌酶谱、凝血功能等。随访至今,病情相对稳定。
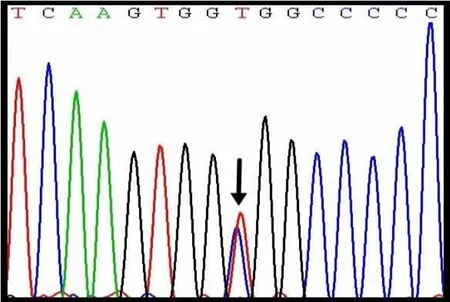
图1 患儿及其父亲、哥哥的基因突变位点
2 讨论
胚胎早期(5~6周)心室肌纤维逐渐致密化,致密化过程为:心外膜→心内膜,心底部→心尖部,疏松的肌小梁形成致密层,较大的梁间隙形成毛细血管网,如果发育过程中发生延迟或停顿,心肌将处于疏松状态,病理表现为粗大的肌小梁及相互交错的深隐窝,称为心肌致密化不全。1932年Bellet等[1]首次对IVNC报道:在1例先天性心脏病患儿尸检中发现其心室肌呈胚胎窦状隙残留。2006年美国心脏病协会(AHA)从基因组及分子定位角度提出新的心肌病定义及分类,将其归为原发性心肌病中的遗传性心肌病。
IVNM发病年龄多见于儿童,临床表现有心功能下降,心律失常及血栓栓塞。该患儿主要表现为呼吸困难、四肢末梢凉等心功能下降表现。有文献报道,18%~40%患者有家族遗传史,多呈常染色体显性遗传[2-3]。部分患儿表现为特殊面容:前额宽,耳际低,颧弓高等。本例患儿无明显特殊面容。
目前超声心动图(UCG)是IVNC主要检查方法,目前多参考Jenny等[4]推荐的成人IVNC诊断标准:(1)不合并其他心脏畸形;(2)具有典型两层不同心肌,即外层的致密心肌和内层的非致密化心肌,其间可见深陷隐窝,收缩末期非致密化与致密化心肌厚度比 >2 0(成人 >2 0,幼儿 >1 4);(3)病变区域主要位于心尖部、侧壁和下壁;(4)彩色多普勒血流显像探及深陷隐窝之间有血流灌注并与心腔相通,而不与冠状动脉循环相通。周佳等[4]曾报道1例胎儿期查心脏超声考虑IVNM,终止妊娠后大体解剖,光镜下见增粗、紊乱肌小梁,内层为非致密层,肌束增大紊乱,外层为致密层,肌束走形、形态整齐、有序,镜下表现与Jenny等[4]超声下诊断标准基本相符。本例患儿经心脏超声诊断,表现为左室壁内膜层不规则增厚,伴左室扩张,心室肌呈“蜂窝样”改变,射血分数降低至44%。由于UCG观察局限性及检查者经验不足等限制,闫朝武等[5]认为心脏磁共振成像(CMRI)因其“一站式”优势可逐渐替代心脏超声,但仍存在争议。
随着二代测序技术成熟,对IVNC基因突变研究逐步增多。目前 IVNC的致病基因有20多种[6-7],一般认为,常染色体显形遗传为主要遗传方式,且该病亦存在遗传异质性,多与导致心肌病、骨骼肌疾病和染色体异常的线粒体相关疾病有关,如首次发现的与X染色体突变有关的Barth综合征(G4 5和TAZ基因突变)、肥厚性心肌病(MYH7、ACTC基因突变)、营养不良蛋白肌病(DTNA突变)等,也有少见基因突变也可引起 IVNC,如LMNA,ZNM9,DMD,PMP22以及本例患儿的MYBPC3。
人MYBPC3基因mRNA全长4 217 bp,位于11号染色体(11p11 2)上,可编码1 274个氨基酸组成的多肽,属于免疫球蛋白超家族,为酸性亲水蛋白,稳定性差,主要二级结构元件是随意卷曲。该基因突变所致家族性肥厚性心肌病(hypertrophic cardiomyopathy,HCM)占42%。MYBPC3基因突变形式复杂:错义突变、插入或缺失、剪接位点突变等,这是区别与其他致病基因的特点。其中缺失突变是该基因常见形式,但本例先证者为错义突变(c 2671C>T)。郑红等[8]发现:移码突变导致的临床表现重于错义突变,特别是有晕厥病史者,是心源性猝死的高危指标。因此,若患儿有晕厥病史,需警惕心源性猝死可能。
MYBPC3编码的肌球蛋白结合蛋白C(cMyBP-C)是一个横纹肌粗肌丝相关蛋白,相对分子质量为140 kDa左右,位于粗肌丝两端肌球蛋白头部横桥处,可与肌动蛋白或肌球蛋白相互作用,捆扎粗肌丝。Winegrad等[9]发现:cMyBP-C不仅是一种结构蛋白,也是一个调节心脏收缩速度及幅度的关键因子,通过磷酸化等调节横桥循环并参与肌肉收缩和舒张。心力衰竭终末期和肥厚性心肌病的特征之一是cMyBP-C磷酸化水平降低[10-11]。根据基因突变位点不同,MYBPC3可引起不同症状,其中LVNC10型的特点是因为心肌形态发育障碍导致左室心肌致密化不全,左心室肥厚,收缩功能差。有研究认为,对小鼠MYBPC3基因治疗可以恢复心肌cMyBP-C水平,并能长期预防心肌病的发生。
对于IVNC的治疗,目前尚无统一治疗方案,多以对症治疗为主。心力衰竭可应用强心剂、利尿剂等治疗,线粒体相关异常可选用维生素B、辅酶Q10及左旋肉碱等治疗,针对心律失常可选用β受体阻滞剂,另外血栓栓塞的预防也很关键。Wang等[12]认为无论是否存在血栓均应长期预防性服用抗凝药物,若出现血栓则需立即溶栓治疗。重症IVNM患儿终末期唯一方案就是心脏移植[13]。
综上,关于IVNC的发病机制仍不清楚,且临床表现缺乏特异性,对于临床医生,应提高对该病认识,以期早期发现,早期干预,延长患儿生存时间。另外,从基因治疗方面,MYBPC3作为IVNC发生发展的主要因素,应是治疗心肌病较好的干预靶点。
猜你喜欢
广西医科大学学报(2022年5期)2022-06-07
世界科学技术-中医药现代化(2022年2期)2022-05-25
昆明医科大学学报(2022年3期)2022-04-19
中国生殖健康(2020年2期)2021-01-18
保健文汇(2020年7期)2020-08-21
保健与生活(2020年13期)2020-07-24
中南医学科学杂志(2019年6期)2019-12-05
中国临床医学影像杂志(2019年1期)2019-04-25
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
