
GC-MS/MS法测定蔬菜中苯醚甲环唑残留量的不确定度评定
2020-06-13 09:17谭锦萍陈羽中黄小清
农产品加工 2020年9期
谭锦萍,阮 征,陈羽中,黄小清
(1.华南理工大学,广东广州 510000;2.广州市食品检验所,广东广州 510410)
苯醚甲环唑又名噁醚唑,是一种具有高效、广谱、低毒和内吸传导性强的三唑类杀菌剂,对作物具有保护和治疗作用,广泛应用于水果和蔬菜等农作物上[1]。通过抑制麦角甾醇的生物合成、破坏细胞膜结构功能,从而达到杀菌目的,对蔬菜和瓜果的纹枯病、锈病、早疫病、叶斑病、黑星病、白粉病等具有较好的保护和治疗作用。由于苯醚甲环唑具有持效期长、与其他杀菌剂无交互抗性、不污染农产品、不杀伤天敌的优点[2-3],加上农民合理使用农药的水平有限、误用和滥用情况严重,容易造成一些病虫害对其产生耐药性。在农产品的日常监督抽检中发现,265个蔬菜样品中检出苯醚甲环唑的阳性样品有50个,检出率高达18.87%。可见,苯醚甲环唑在蔬菜种植中比较常用。
标准[4]规定,某些蔬菜苯醚甲环唑的最高残留限量 (Maximum residue limit,MRL) 为 0.2 mg/kg。在实际检测中,当苯醚甲环唑的检出值在限量值附近时,实验室出具的检测结果直接关系到样品是否合格的判定。因此,为确保结果的可信度,必须进行不确定度的评估[5]。
1 材料与方法
1.1 材料仪器与试剂
蔬菜,购于广州某超市。
TSQ8000evo型气相色谱-三重四极杆质谱仪、EI离子源,美国Thermo Scientific公司产品;BP221S型电子天平,美国赛多利斯公司产品;2-16KL型高速离心机,德国Sigma公司产品;HS501型数显往复振荡摇床,德国IKA仪器设备有限公司产品。
苯醚甲环唑(100 μg/mL),农业部环境保护科研检测所提供;环氧七氯、乙腈(色谱纯),美国Fisher公司提供;QuEChERS分散固相萃取盐包(内含4 g MgSO4、1 g NaCl、1 g柠檬酸钠、0.5 g三水合二柠檬酸二钠)、QuEChERS分散固相萃取净化管(150 mg PSA和900 mg MgSO4)、陶瓷均质器,美国Agilent公司提供。
1.2 试验条件
1.2.1 色谱条件
色谱柱:TR-PESTICIDEⅡ毛细管柱(30 m×0.25 mm,0.25 μm);初始柱温40℃,保持1.5 min,以 25 min/℃升至90℃,保持1.5 min,以25 min/℃升至180℃,以5℃/min升至280℃,再以10 min/℃升至300℃,保持5 min;进样口温度270℃,进样量1 μL,不分流;载气为高纯氦气,1.2 mL/min。
1.2.2 质谱条件
离子源温度300℃,传输线温度300℃,EI离子源70 eV。
定性、定量离子和碰撞能见表1。

表1 定性、定量离子和碰撞能
1.2.3 标准储备液及内标溶液的配制
准确称取25 mg环氧七氯标准物质,用正己烷溶解并定容至25 mL,得到1 mg/mL的溶液,作为内标的标准储备液;准确吸取200 μL的内标标准储备液,正己烷定容至10 mL,得到20 mg/L的内标工作溶液。精确吸取苯醚甲环唑标准溶液1 mL,用乙腈溶解并定容至10 mL,得到10 mg/L的标准工作溶液。
1.2.4 标准曲线的制备
准确吸取 0.05,0.10,0.20,0.50,1.00,2.00 mL的标准溶液于10 mL容量瓶中,准确各加入50 μL的20 mg/L内标工作溶液,用蔬菜的基质空白溶液定容。以苯醚甲环唑浓度与内标物浓度的比值(C苯醚甲环唑/C内标)为横坐标、苯醚甲环唑峰面积与内标物峰面积(A苯醚甲环唑/A内标) 的比值为纵坐标,绘制标准曲线。
1.2.5 前处理方法
(1) 提取。均匀取样并准确称取样品10 g(精确到0.01 g),置于50 mL离心管中, 加入20 mg/L的内标工作溶液100 μL,加入20 mL的乙腈,于组织捣碎机上提取2 min,置于-18℃冰箱中冷冻10 min后取出,加入QuEChERS萃取包、2粒均质子,剧烈振摇1 min后,以转速8 000 r/min离心3 min。
(2) 净化。分取上清液 5 mL于QuEChERS净化管中,剧烈振摇1 min后,以转速8 000 r/min离心3 min,上清液过滤膜后,用气相色谱-三重四极杆质谱仪测定。以保留时间和离子对丰度比定性,内标曲线法定量。
1.2.6 回收率的测定
准确称取各10.00 g空白蔬菜样品6份,于50 mL离心管中,加入相同质量浓度苯醚甲环唑标准和内标,按1.2.5前处理方法同步进行,测定苯醚甲环唑的含量,并对其回收率和精密度进行计算。
2 结果与分析
2.1 数学模型的建立
从测定苯醚甲环唑的数学模型中,分析其不确定度的来源。就试验而言,采用内标法计算样品中苯醚甲环唑残留量的计算公式为:

式中:X——样品中苯醚甲环唑的含量,mg/kg;
C——由A样/A内标从曲线中得出的试样中苯醚甲环唑的质量浓度,mg/L;
V——提取液体积,mL;
m——称量质量,g;
frec——加标试验的回收率,%;
根据JJF 1059—1999[6],由相对标准不确定度合成标准不确定度,合成标准不确定度公式为:

2.2 试验结果
取6个加标的蔬菜样品,按照1.2.5的处理方法对蔬菜样品中苯醚甲环唑进行平行6次的试验,对其含量进行测定。
测定结果见表1。

表1 测定结果
由表1可以计算出本次试验测定的苯醚甲环唑含量为:
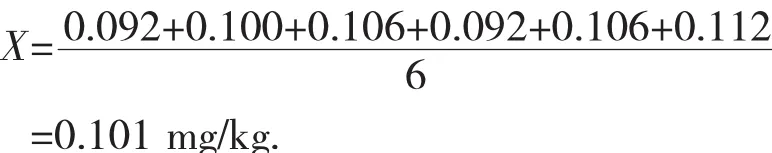
3 不确定度评定
3.1 不确定度的来源分析
根据内标法原理,被测样品和标准系列溶液中加入的内标物相同,即内标物的浓度可约除,因此无需考虑内标物纯度引起的不确定度,只考虑内标物加入的体积。纵观整个检测过程,对蔬菜中苯醚甲环唑残量结果有影响的不确定度分量来源有标准物质、样品、样品前处理过程、仪器分析过程等。
3.2 标准物质引起的不确定度Urel(标)
3.2.1 标准物质浓度引起的不确定度Urel(标p)
苯醚甲环唑标准溶液购自农业部环境保护科研检测所,根据证书信息,苯醚甲环唑的标准值为100 μg/mL,扩展不确定度为 0.19 μg/mL。

3.2.2 标准溶液配制过程引起的不确定度Urel(标v)
试验的标准储备液的配制过程使用了10 mL的容量瓶(A级),标准曲线配制过程使用了100 μL和1 mL移液器,按照JJG 196—2006《常用玻璃量器检定规程》[7]和JJG 646—2006《移液器》[8]检定规程,玻璃器和移液器均有相应的最大允许误差,其不确定来源包括以下2个方面。
(1)容量瓶、移液器本身的不确定度。检定证书指出10 mL容量瓶(A级)的最大允差为±0.020 mL,100 μL移液器的最大允差为±2.0%,1 mL移液器最大允差为±1.0%,由量具允差引入的相对标准不确定度公式为:

(2)环境温度引起的不确定度。实验室的温度一般控制在20±5℃,20℃时乙腈的膨胀系数是1.37×10-3℃,由环境温度引起的相对不确定度公式为:
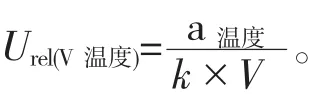
根据各分量合成标准溶液的配制过程中引入的相对不确定度为:

玻璃器具和移液器的相对标准不确定度见表2。
3.2.3 标准校正曲线拟合的不确定度Urel(标w)
试验对6个不同浓度的苯醚甲环唑进行测定。内标校正曲线见表3。

表2 玻璃器具和移液器的相对标准不确定度

表3 内标校正曲线
以系列标准溶液中苯醚甲环唑与内标物质量浓度比值为横坐标,苯醚甲环唑与内标物丰度比值为纵坐标,得到的回归方程为Y=4.618X-0.193 1,R2=0.999 3。
计算标准溶液待测物质信号残差的标准差为:

对苯醚甲环唑为阴性的蔬菜样品进行5次的添加回收试验(添加量为0.2 mg/kg)。
样品测定结果(n=5) 见表4。

表4 样品测定结果(n=5)
校准曲线拟合的标准不确定度为:

式中:S——标准溶液待测物质信号残差的标准差,0.1;
b——标准曲线的斜率,4.618;
P——试验次数,5;
n——标准曲线的浓度点数,6;
C0——样品中苯醚甲环唑的实际测定浓度,0.105 2;
c¯——各标准溶液浓度平均值,0.64;
Sn——所有校准溶液浓度的离均差平方和。
则曲线拟合的引入的相对不确定度为Urel(标w)=U(std)/1.052=0.084/1.052=0.079 8.
综上分析,根据标准品物质所引入的不确定度来源,计算由标准物质引入的相对标准合成不确定度为:
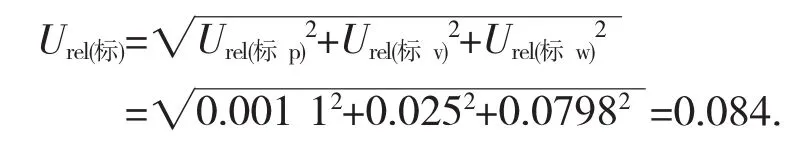
3.3 仪器重复测量引入的不确定度Urel(仪)
仪器引入的不确定度是由仪器对被测组分的响应值(峰面积)的重复性差异引起的,按A本类评定,可通过重复进样来计算。通过将同一样品溶液进行连续6次重复进样,得到的重复进样结果。
重复进样测定结果见表5。
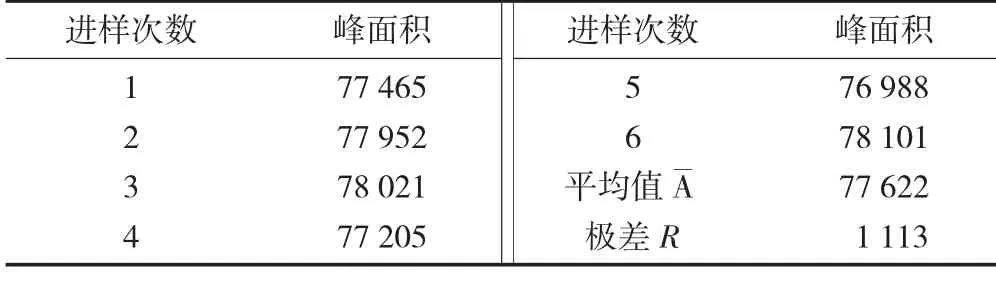
表5 重复进样测定结果
根据标准[6],在测定次数少的情况下,可采用极差法对不确定度进行评定,查表可得,当n=6,C=2.53。即由重复性进样引入的标准不确定度和相对标准不确定度为:
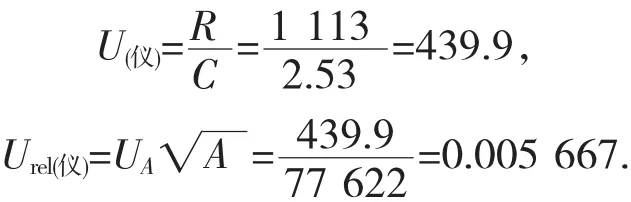
3.4 样品前处理中引起的不确定度Urel(样)
样品处理过程不确定度的主要来源:一是样品使用的称量天平引起的不确定度;二是样品提取、净化、定容等过程中所使用的玻璃量器所引起的不确定度。
样品用百分之一天平进行称量,其校准允差为±0.01g,试验称取样品量为10 g,k按均匀分布计算,K取;样品净化、定容过程中使用的是20 mL的移液管。
天平和玻璃量器引起的不确定度见表6。
由表6可知,样品前处理过程所引起的相对标准不确定度合成为:

3.5 结果计算过程引入的不确定度Urel(frec)
回收率是结果计算中的一个重要的不确定度来源因子。取低、中、高3个水平进行苯醚甲环唑的添加回收试验,每个水平进行6次试验,按照2.2.5步骤进行处理上机。对18次的测定结果进行添加回收率计算。

表6 天平和玻璃量器引起的不确定度
回收率结果统计见表7。

表7 回收率结果统计
回收率按A类不确定度进行评定计算,当n=18,
则,由回收率引入的标准不确定度为:
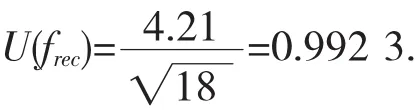
由回收率引入的相对标准不确定度为:
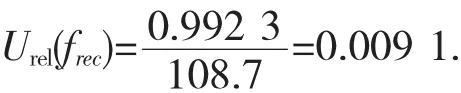
4 不确定度的合成及扩展
4.1 各相对标准不确定度分量的合成
各相对标准不确定度分量见表8。

表8 各相对标准不确定度分量
苯醚甲环唑的相对合成不确定度为:

折算合成标准不确定度为:

4.2 扩展不确定度的评定
取置信水平为95%,k=2,计算扩展不确定度U(C)=u(C)×k=0.017×2=0.034。因此,样品中苯醚甲环唑的残留量表示为(0.2±0.0.034) mg/kg(k=2)。
5 结论
以苯醚甲环唑为主要研究对象,对蔬菜中苯醚甲环唑的不确定度评估进行了论述。当取样量为10 g,k=2(95%置信度) 测得的蔬菜中苯醚甲环唑的含量为(0.2±0.034) mg/kg。表明整体方法的准确性较好,可用于蔬菜中苯醚甲环唑的测定和不确定评估。
猜你喜欢
现代农药(2022年4期)2022-08-12
现代农村科技(2022年3期)2022-03-14
口腔护理用品工业(2021年4期)2021-11-02
科技创新导报(2020年5期)2020-06-11
中国科技纵横(2019年23期)2019-02-14
植物保护(2018年3期)2018-05-14
科教导刊(2017年26期)2017-11-07
海峡科技与产业(2017年1期)2017-03-04
科技与创新(2015年17期)2015-09-11
科技与创新(2014年12期)2014-08-28
