
环硼氧烷与碱金属形成复合物的密度泛函理论研究
2020-06-12 05:19:02徐秋红
山东化工 2020年8期
徐秋红
(滨州学院 化工与安全学院,山东 滨州 256603)
环硼氧烷(B3O3H3)作为苯的等电子体,是由硼氧原子交替形成的平面六元环结构,俗称“无机苯”。无机苯在材料科学领域中有着越来越重要的应用前景,作为配体形成的化合物往往在光学、电学、磁学等方面表现出优异的性质。环硼氧烷与Cu、Ag、Au等过渡金属相互作用形成半夹心、夹心型复合物已有过相关研究[1],而碱金属作为配体形成的复合物也有过相关研究[2-3]。据我们所知,环硼氧烷与碱金属形成复合物的研究较少,本文运用Gaussian16软件[4],采用密度泛函理论研究了环硼氧烷与碱金属阳离子Li+、Na+、K+的相互作用,进行成键特征分析,寻找最稳定结构,以期为环硼氧烷的应用提供借鉴和参考。
1 计算方法
采用密度泛函理论的B3LYP和PBE1PBE方法,6-311+G(d,p)基组对(B3O3H3)nM+(M=Li、Na、K;n=1,2)复合物进行优化和频率计算。两种方法得到的结构是一致的,只是异构体间的键长和相对能量略有不同。本研究主要采用B3LYP方法的计算结果,异构体中所有的能量均采用零点校正能(ZEP)。使用NBO5.0[5]程序计算键级和自然键电荷,用Origin软件模拟其红外吸收光谱(IR)。
2 结果与讨论
2.1 几何结构
图1列出了用B3LYP和PBE1PBE方法优化得到的B3O3H3和C6H6的结构。从图1可以看出,B3O3H3具有D3h对称性,1A1′电子态,而C6H6具有D6h对称性,1A1g电子态,B3O3H3中B-O键长为1.39 Å,B-H键长为1.17 Å,C6H6中C-C键长1.41 Å,C-H键长1.10 Å,与文献报道的一致[6],说明了用B3LYP和PBE1PBE计算方法的正确性。
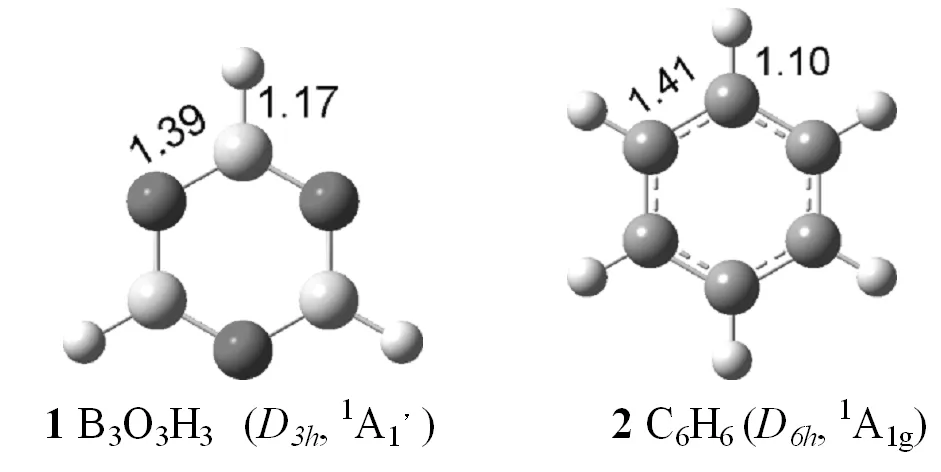
图1 优化的B3O3H3和 C6H6的最稳定结构,键长的单位为ÅFig.1 Low-lying isomers of B3O3H3 and C6H6calculated by B3LYP level,bond lengths in Å
B3O3H3M+(M=Li、Na、K)复合物的最稳定结构,如图2所示。由图2可以看出,B3O3H3Li+、B3O3H3Na+和B3O3H3K+的最稳定结构都为平面型结构,具有C2v对称性,1A1电子态。而文献报道的苯与碱金属离子形成的最稳定结构是半三明治结构[7-8]。B3O3H3M+(M=Li、Na、K)复合物的最稳定结构中,金属离子Li+、Na+和K+都是与B3O3H3中的O原子相互作用,从表1可以看出,金属离子都带有正电,分别为0.633、0.854和0.828|e|,而O都带有负电,分别为-0.259、-0.321、-0.282|e|。 复合物中,O- M+(M=Li、Na、K)键长,随着离子半径的增加而增加,O-Li+、O-Na+和O-K+键长分别为1.74、1.79和2.50 Å。表1给出了复合物的wiberg键级,O-Li+、O-Na+和O-K+的键级分别为0.265、0.106和0.142,从键级可以看出,B3O3H3Li+复合物的键级最大,结合最稳定。表1给出了复合物中金属离子的离子键成分,可以看出,三者的离子键成分分别为0.95、0.97、0.95,说明B3O3H3和M+(M=Li、Na、K)的相互作用主要是离子键作用。

图2 优化的B3O3H3M+ (M=Li、Na、K)的最稳定结构,键长的单位为ÅFig.2 Low-lying isomers of B3O3H3M+(M=Li,Na,K) calculated by B3LYP level,bond lengths in Å

表1 基于B3LYP/6-311+G(d,p)水平下基态结构的自然键电荷q/| e |和wiberg键级Table 1 Calculated natural atomic charges(q/|e|),wiberg bond indexes(WBI) of the ground staes of B3O3H3M+ (M=Li,Na,K) at B3LYP/6-311+G(d,p) level.
图3给出了优化的(B3O3H3)2M+(M=Li、Na、K)复合物的最稳定结构。有趣的是,(B3O3H3)2Li+、(B3O3H3)2Na+和(B3O3H3)2K+的最稳定结构和第二稳定结构中,两个B3O3H3都是通过中间的金属离子连接而成的,不同是两个B3O3H3的连接方式,前者为垂直连接,后者为水平连接。更有趣的是,(B3O3H3)2Li+、(B3O3H3)2Na+和(B3O3H3)2K+的最稳定结构都具有D2d对称性,1A1电子态,而第二稳定结构都具有D2h对称性,1Ag电子态。不仅如此,(B3O3H3)2Li+、(B3O3H3)2Na+和(B3O3H3)2K+最稳定结构和第二稳定结构中,O-M+(M=Li、Na、K)的键长、B-O键长、B-H键长,都分别相等。在B3LYP水平下,(B3O3H3)2Li+、(B3O3H3)2Na+和(B3O3H3)2K+最稳定结构比第二稳定结构分别高0.61、0.04和0.02 kal·mol-1,能量相差特别小,说明二者在实验上是共存的。(B3O3H3)2M+(M=Li、Na、K)复合物中,O-Li+、O-Na+和O-K+键长随着离子半径的增加而增加。对比(B3O3H3)2M+和B3O3H3M+(M=Li、Na、K)复合物,发现O-Li+(1.75和1.74Å)和O-K+(2.51和2.50 Å)键长分别近似相等,而B3O3H3Na+和(B3O3H3)2Na+复合物中,O-Na+键长变化比较明显,从1.79 Å变化到2.04Å。
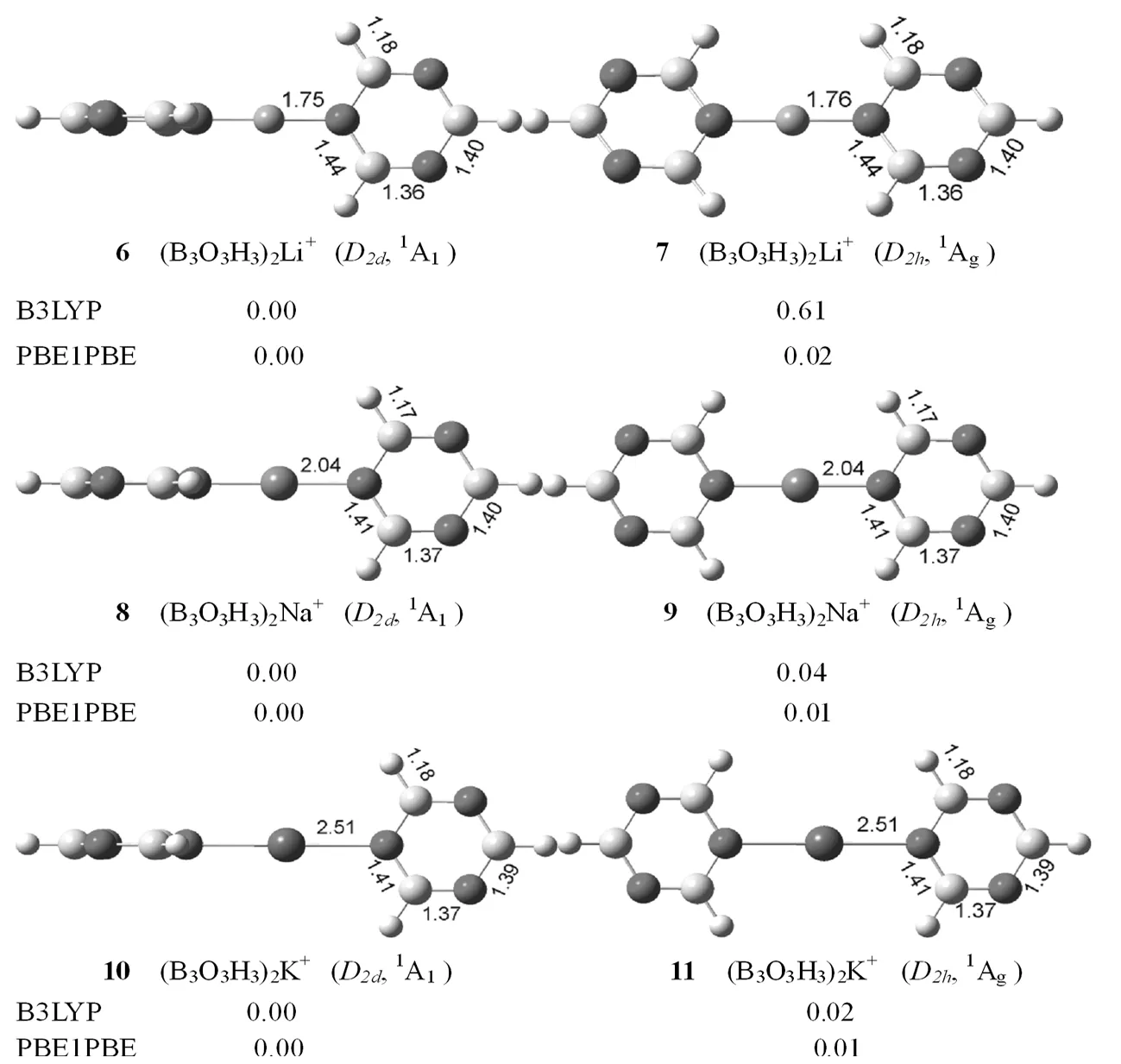
图3 优化的(B3O3H3)2M+ (M=Li,Na,K)的稳定结构,键长的单位为ÅFig.3 Low-lying isomers of (B3O3H3)2M+ (M=Li、Na、K) calculated by B3LYP level,bond lengths in Å
3.2 红外分析
图4分别给出了B3O3H3M+和(B3O3H3)2M+(M=Li,Na,K)复合物基态结构的红外吸收光谱。B3O3H3中913.67和1300.89 cm-1为B-H的摇摆振动,1503.80 cm-1为B-O的伸缩振动,3509.44 cm-1为B-H的伸缩振动。B3O3H3M+(M=Li,Na,K)中,B-H的摇摆振动分别在1406.12、1437.53和1444.67 cm-1,而B-O的伸缩振动分别在1552.79、1551.96和1541.88 cm-1,相对于B3O3H3,向高波数方向移动,即发生了蓝移。对于(B3O3H3)2M+(M=Li,Na,K)复合物,B-H的摇摆振动分别在1417.87、1443.56和1448.98 cm-1,而B-O的伸缩振动分别在1538.62、1543.22和1535.50 cm-1,相对于B3O3H3,向高波数方向移动,即发生了蓝移。
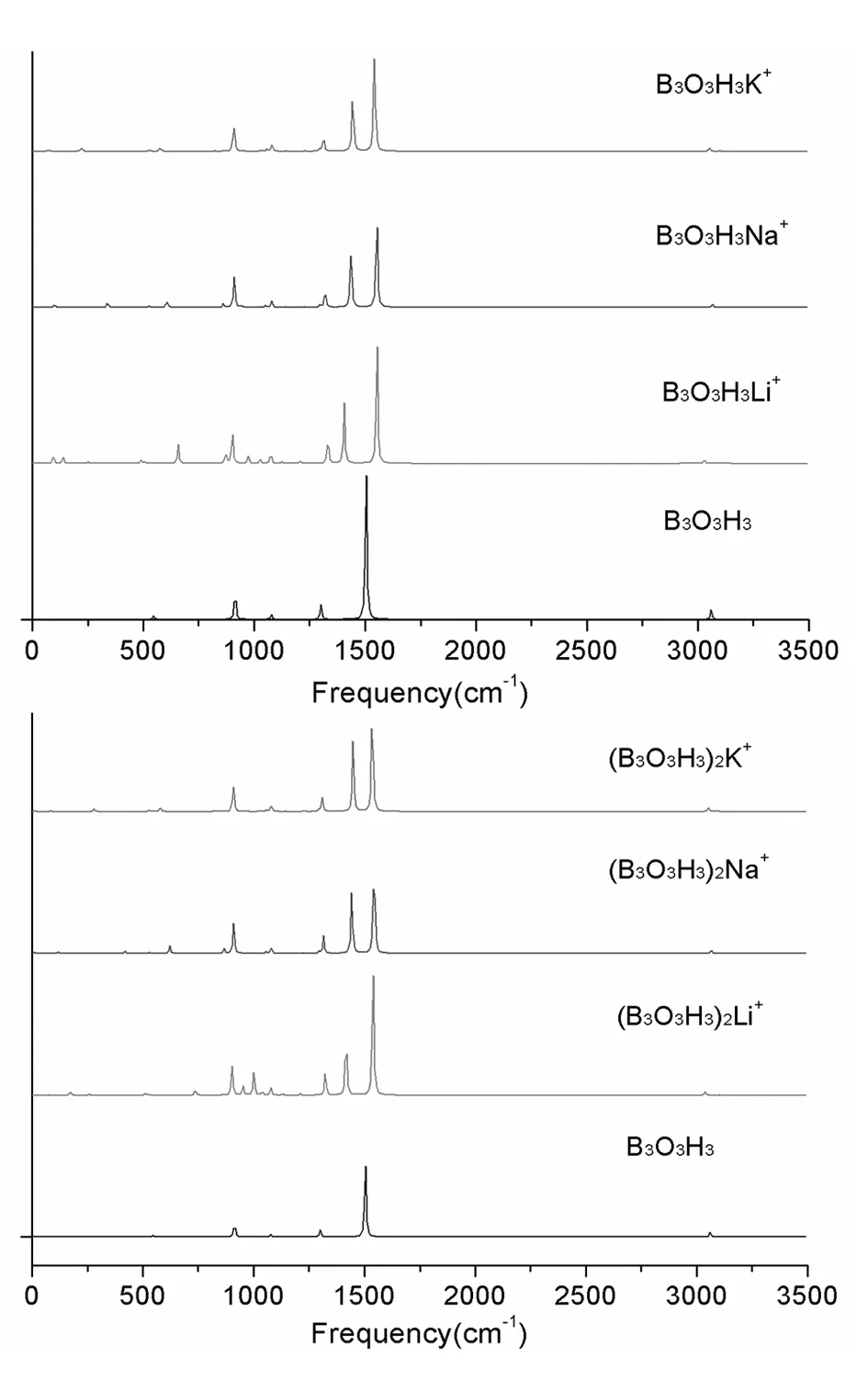
图4 B3O3H3M+和(B3O3H3)2M+(M=Li,Na,K)基态结构的红外吸收光谱Fig.4 IR spectra of the ground states of B3O3H3M+and (B3O3H3)2M+(M=Li,Na,K)
4 结论
基于密度泛函理论的B3LYP和PBE1PBE方法确定了B3O3H3M+和(B3O3H3)2M+(M=Li,Na,K)复合物的基态结构。结果发现,基态结构中,金属离子倾向于与B3O3H3中的O相互作用,B3O3H3M+具有C2v对称性,而(B3O3H3)2M+(M=Li,Na,K)具有D2d对称性。B3O3H3M+基态结构中,金属离子与B3O3H3的相互作用主要是离子键作用。在红外吸收光谱中,最大的峰为环上B-O的伸缩振动峰,B-H的摇摆振动和B-O的伸缩振动分别发生了蓝移。
猜你喜欢
数学物理学报(2022年3期)2022-05-25 13:33:22
数学物理学报(2022年1期)2022-03-16 06:15:04
数学物理学报(2021年5期)2021-11-19 07:01:16
数学物理学报(2021年3期)2021-07-19 06:02:18
高中数理化(2020年16期)2020-10-14 11:51:52
考试周刊(2019年2期)2019-01-28 10:08:56
中学化学(2015年5期)2015-07-13 07:41:41
中学化学(2015年5期)2015-07-13 07:36:59
湖南师范大学自然科学学报(2015年1期)2015-02-27 14:50:04
化学教与学(2014年12期)2014-12-12 10:05:09
