
氧化铝负载的钴基费托合成催化剂失活机理
2020-05-20 06:57秦绍东李加波何若南段雪成孟祥堃
煤炭学报 2020年4期
秦绍东,李加波,何若南,杨 霞,段雪成,孟祥堃
(北京低碳清洁能源研究院,北京 102211)
费托合成(FTS)是将合成气转化为燃料与化学品的重要工业路线之一。已经工业化的费托合成催化剂有Fe基与Co基两类,目前使用Fe基催化剂的费托合成技术已经在国内实现大规模工业应用,而使用Co基催化剂的费托合成技术尚不成熟。与Fe基相比,Co基催化剂具有费托合成活性高,重质烃选择性高与水煤气变换(WGS)活性低的优势,但劣势是催化剂的生产成本高与耐硫性差。
为提升Co基费托合成技术的经济性与竞争力,提高Co基催化剂的稳定性,延长其使用寿命尤为重要。而提升稳定性的前提是需要对Co基催化剂在费托合成反应中的失活机理有清晰的认识。文献中对Co基催化剂的失活机理有大量研究[1-5]。金属Co活性相的氧化曾一度被广泛认为是Co基催化剂的主要失活原因[3,6],但近期Sasol公司的研究者认为当金属Co的晶粒尺寸在4.5 nm以上时,负载型的金属Co在真实的费托合成气氛中不会被氧化[3,7]。催化剂表面的积碳是另一种被广泛认可的Co基催化剂失活机理,借助程序升温氧化(TPO)与程序升温加氢(TPH)分析,研究者证实反应后催化剂表面有大量的积碳生成[2,5]。除了金属Co的氧化与碳沉积,金属Co烧结被认为是催化剂失活的另一重要原因。反应后工业Co基催化剂的透射电子显微镜(TEM)分析证实催化剂中金属Co的晶粒尺寸在初始反应的一段时间内会快速增加,然后逐渐保持稳定[7]。
对于采用SiO2与Al2O3等金属氧化物作为载体负载的催化剂,Co与载体之间的相互作用是不可避免的,在费托合成反应中Co与载体反应生成新的化合物(如Co的硅酸盐与铝酸盐)也被认为是催化剂失活的重要原因[8-9]。对于Co/Al2O3催化剂,由于铝酸钴在真实的费托合成反应条件下较难生成,因此近期部分研究人员认为铝酸钴不是催化剂失活主要原因[10]。在费托合成反应过程中金属氧化物载体,如SiO2,还会与水蒸气反应生成硅的水合物,也被证实会导致Co/SiO2催化剂的失活[8]。
在工业反应中,费托合成的原料气中不可避免的含有微量的硫、氮等对催化剂有毒害作用的杂质,因此硫化物与氮化物导致的催化剂中毒是工业催化剂失活的重要原因[11-14],但在实验室中催化剂的硫化物与氮化物中毒可采用控制气体纯度等手段避免。
本研究采用浸渍法制备了Co/Al2O3催化剂,在典型工业费托合成反应条件下,采用搅拌釜反应器对该催化剂反应性能进行评价。通过对不同反应时间催化剂的系统表征,获取Co/Al2O3催化剂失活机理,进而为后期高稳定性Co基费托合成催化剂的开发提供指导。
1 实 验
Co/Al2O3催化剂采用两步浸渍法制备。具体方法如下:室温下配制一定浓度的Co(NO3)2溶液,然后向该溶液中加入定量的γ-Al2O3载体进行等体积浸渍,在80 ℃水浴中将样品蒸干后,于120 ℃干燥脱水。将干燥后的样品按照上述步骤二次浸渍Co(NO3)2溶液,120 ℃再次干燥后,于马弗炉中焙烧得到催化剂。
催化剂的比表面积和孔结构在Micromeritics ASAP 3020物理吸附仪上测定,测试前催化剂在90和250 ℃脱气6 h。X射线粉末衍射(XRD)分析在日本理学Dmax2600型X射线粉末衍射仪上进行,Cu Kα靶,电压40 kV,电流100 mA,扫描范围2θ=15°~70°。TPH在Micromeritics Auto chem.II 2920上进行,装置配有小型质谱检测仪,测试中取100 mg样品放入U形管中,在50 mL/min的10% H2/90% Ar混合气中程序升温,升温速率为10 ℃/min,通过小型质谱仪测得尾气的CH4信号(M/Z=15)变化。TEM图片采用JEOL 2011电子显微镜在200 kV下测得。
催化剂评价在300 mL搅拌釜反应器中进行,催化剂装填量1.0 g。反应前称取1.0 g催化剂粉末装入固定床反应器中进行活化处理,反应器入口与出口均安装有截止球阀。活化在高纯H2气氛中,375 ℃,0.1 MPa进行10 h,活化完成后,将反应管进口与出口的球阀封闭,然后转入N2气氛的手套箱中,在手套箱中将反应管中活化后的催化剂转移至装有液体石蜡的搅拌釜反应器中。费托合成性能测试在H2/CO=2.0(体积比)的合成气中,于220 ℃,2.0 MPa,10 000 h-1下进行,反应中通入一定量的N2用作内标气。为避免催化剂硫中毒的影响,催化剂性能测试中所使用的气体均为高纯气。反应钢瓶气体经过减压阀减压后先依次经过两个装有脱硫剂与脱水剂的罐体进行气体纯化处理,净化后的气体经布鲁克斯质量流量计控制流量后进入搅拌釜反应器。反应中产物依次经过热井(140 ℃)与冷井(0 ℃)后与气相尾气分离。反应尾气中CO,CO2,CH4,C2H4与C2H6等物种的含量利用在线的安捷伦7890A气相色谱(配备TCD+FID检测器)进行分析。
用于表征分析的反应后催化剂样品,除XRD外,其余在表征分析前均先用甲苯在手套箱N2气氛中80 ℃进行多次抽提处理,目的是去除反应过程中残留在催化剂表面的烃类,抽提后的催化剂样品密封在乙醇溶液中,然后用于BET,TPH与TEM表征分析。
2 结果与讨论
2.1 催化剂的费托合成性能
Co/Al2O3催化剂的费托合成性能在搅拌釜反应器中,在合成气H2/CO=2.0(体积比),220 ℃,2.0 MPa,10 000 h-1条件下进行测试。催化剂的CO转化率与CH4选择性随反应时间的变化结果如图1所示,可见在整个费托合成性能测试中,随着反应时间的延长催化剂CO转化率逐渐降低,CH4选择性缓慢升高。这表明费托合成催化剂在持续性的失活,该催化剂的稳定性结果与文献中报道的工业Co/Al2O3催化剂的初期反应性能一致[7]。

图1 催化剂CO转化率与CH4选择性随反应时间变化Fig.1 Changes of CO conversion and CH4 selectivity with reaction time
为研究催化剂的失活机理,对制备的Co/Al2O3催化剂分别进行不同反应时间的费托合成性能测试,测试后的催化剂从反应釜中取出后进行表征分析。
2.2 催化剂织构性质变化
表1为反应35 h与反应160 h 后的催化剂的织构性质结果。可见,随着反应时间的增加催化剂的比表面积,孔容与平均孔径均有增加的趋势,但增加量均在物理吸附的误差范围内(<10%),因此可以认为催化剂比表面积,孔容与平均孔径随着反应时间的延长无显著变化。不论是催化剂的比表面积,孔容与平均孔径随反应时间缓慢增加,还是变化不大,均可证明催化剂失活不是催化剂比表面积与孔结构变化所致。
表1 不同反应时间的催化剂织构性质
Table 1 Structural properties of catalysts with different reaction times
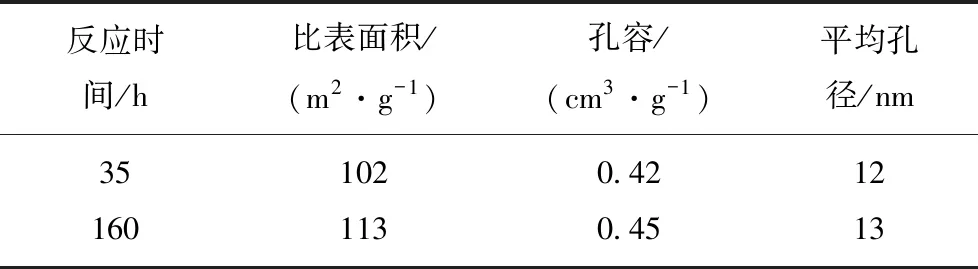
反应时间/h比表面积/(m2·g-1)孔容/(cm3·g-1)平均孔径/nm351020.42121601130.4513
2.3 物相与晶粒变化
图2为反应不同时间的催化剂XRD谱图。反应前催化剂(0 h)的XRD谱图中可以观察到金属Co,CoO与γ-Al2O3的衍射峰。随着反应的进行,CoO的衍射峰快速减弱并消失,表明反应过程中CoO被进一步还原为金属Co。与此同时,金属Co的衍射峰强度缓慢增加,表明金属Co的晶粒尺寸随着反应的进行在逐渐增大,即催化剂中的活性相金属Co在逐渐发生烧结。

图2 不同反应时间催化剂的XRD谱图Fig.2 XRD patterns of catalysts with different reaction times
Co/Al2O3催化剂不同反应时间的XRD表征结果证实在费托合成反应中金属Co没有被氧化,相反,未被还原的CoO在反应气氛中被逐渐还原为金属Co,相似的结果在其他文献也有报道[14]。上述结果表明在本研究中活性相金属Co的氧化不是导致催化剂失活的原因。
STEEN等[16]通过热力学分析研究了不同晶粒尺寸的纳米金属Co在水与氢气混合气氛中的氧化与还原的稳定性,其结果表明在真实的费托反应条件下晶粒小于4.4 nm的球形金属Co易于被氧化,而大于4.4 nm的球形金属Co不会被氧化。SAIB等[15]采用X射线吸收近边缘光谱(XANES)研究了100桶/d的中试反应器中Co/Pt/Al2O3催化剂的物相变化,通过对不同反应时间的催化剂分析后证实催化剂中尺寸晶粒>6 nm金属钴具有较好的抗氧化性,在真实的费托合成反应过程中这些金属Co不会被费托合成反应中生成的水蒸气氧化。
在本研究中,通过TEM图片(图3)可见,Co/Al2O3催化剂中金属Co以球形存在,球形金属Co晶粒尺寸绝大多数在6 nm以上,因此在费托合成反应中,催化剂中的金属Co较为稳定,不易于被氧化,这与XRD结果相一致。

图3 不同反应时间催化剂的TEM暗场图片Fig.3 TEM dark-field images of catalysts with different reaction times
XRD表征还证实随着反应的进行催化剂中活性相金属Co在发生烧结,因此可以得出结论:活性相金属Co的烧结是催化剂失活的重要原因。 为进一步证实该结论,使用TEM进一步对催化剂进行表征。
图3为不同反应时间催化剂的TEM暗场图片。图片中明亮的圆形或椭圆形的球为金属Co的颗粒,对比不同反应时间催化剂TEM图片可见,随着反应时间的延长,金属Co颗粒尺寸逐渐增加,这与XRD结果一致,进一步证实在反应过程中活性相金属Co在发生烧结。
2.4 积碳研究
图4为不同反应时间催化剂的TPH谱图。谱图根据出峰温度可划分为两个区域,结合文献报道[2,5,17],500 ℃之前的峰对应于费托合成反应中催化剂表面生成的烃类残留,而500 ℃后的峰对应的是高聚合度的碳物种。在费托合成反应中,CO在催化剂表面发生解离并形成表面碳化物是费托合成反应中的基元反应步骤,这些碳化物可以进一步加氢转化为烃类,也可能转化为不活泼的碳物种,如聚合碳或石墨碳。不活泼的碳物种的生成反应是不可逆的,随反应的进行不活泼的碳物种可以发生持续累积,进而对费托合成反应产生不利的影响。MOODLEY等[2]的研究表明费托合成反应中生成的不活泼的聚合碳主要位于Co/Pt/Al2O3催化剂的金属Co表面与氧化铝载体的孔道中。在本研究中通过图4可知,随着反应时间的延长,催化剂表面不可逆的高聚合度碳物种的量快速增加,这表明催化剂表面的积碳在加剧。显然,在本研究中积碳是导致Co/Al2O3催化剂失活的另一重要原因。

图4 不同反应时间催化剂的TPH谱图Fig.4 TPH spectra of catalysts with different reaction times
3 改进后催化剂稳定性测试
通过上述对不同反应时间Co/Al2O3催化剂的表征可以推断金属Co的烧结与催化剂的积碳是催化剂失活的重要原因。我们从孔结构可控的Al2O3载体开发入手,通过调控Al2O3载体孔径尺寸与分布实现其负载的金属Co的晶粒尺寸与晶粒分布的控制。图5为调控前与调控后催化剂中金属钴晶粒分布的变化,可见调控后催化剂中小于5 nm的金属钴颗粒占比显著减少。通过减少催化剂中易烧结的小晶粒金属Co的生成量,催化剂的抗烧结性能被显著改进。与此同时,通过对催化剂配方的改进,成功开发了具有高抗烧结性与抗积碳能力的Co基费托合成催化剂。催化剂在搅拌釜反应器中,合成气H2/CO(体积比)=2.0,2.0 MPa,10 000 h-1反应条件下进行了长周期稳定性测试,图6为催化剂性能测试结果。在前1 050 h催化剂的测试温度为225 ℃,此时催化剂CO转化率维持在55%左右,CH4选择性维持在6%以下,在整个测试中催化剂费托合成性能保持稳定。1 050 h后将反应温度提升至230 ℃,目的是通过提升催化剂的转化率让催化剂加速失活。升温后催化剂CO转化率升高至约65%,CH4选择性基本不变。在随后的反应中催化剂活性缓慢降低,1 600 h后催化剂的CO转化率降低至63%然后保持稳定。图6中稳定性测试结果证实我们开发的Al2O3负载的Co基费托合成催化剂具有较好的稳定性。与文献中报道的同类催化剂相比[7,18-19],该催化剂避免了需要较长的诱导期才能达到稳态的缺点,因此在后期工业应用中具有显著优势。

图5 催化剂金属钴晶粒分布Fig.5 Co metal particle size distribution of catalyst

图6 优化后催化剂稳定性测试Fig.6 Stability test of the optimized catalyst
文献报道中虽然铝酸钴的生成也被认为是Co/Al2O3催化剂失活原因[6,20],但在真实费托合成反应中该物相很难被定量与表征。考虑到该物相更易于在高水蒸气分压气氛下生成,而在真实费托反应条件下即使该物相生成含量也相对较少[10],因此该物相不应该被认为是Co/Al2O3催化剂失活的主要原因。笔者以改进催化剂抗烧结与抗积碳能力作为出发点,成功开发了具有高稳定性的Co/Al2O3费托合成催化剂,进一步支持了烧结与积碳为Co/Al2O3催化剂在费托合成反应中主要失活原因的结论。
4 结 论
(1)在搅拌釜反应器中对浸渍法制备的典型Co/Al2O3催化剂的费托合成性能进行了测试,发现在整个测试过程中催化剂持续失活。
(2)采用不同的表征手段对不同反应时间的催化剂的织构性质与物化性质进行了表征,表征结果证实烧结与积碳是Co/Al2O3催化剂失活主要原因。
(3)通过对催化剂载体孔结构控制及催化剂配方的改进,提升了催化剂抗烧结性与抗积碳能力,改进后的催化剂在1 800 h的搅拌釜测试中表现了较好的稳定性。
猜你喜欢
科学导报(2022年28期)2022-05-24
科学与财富(2021年33期)2021-05-10
发明与创新·大科技(2019年6期)2019-09-06
汽车零部件(2018年5期)2018-06-13
中南大学学报(自然科学版)(2015年1期)2015-09-22
同济大学学报(自然科学版)(2013年3期)2013-03-04
中国乡村医药(2011年10期)2011-08-30
中国循证儿科杂志(2011年6期)2011-01-19
