
热带假丝酵母磷酸甘油酸激酶基因启动子功能鉴定
2020-05-19 02:03周静雨陈献忠张利华
食品与生物技术学报 2020年3期
周静雨, 陈献忠*, 张利华, 沈 微, 樊 游
(1. 江南大学 工业生物技术教育部重点实验室,江苏 无锡214122;2. 江南大学 生物工程学院,江苏 无锡214122)
启动子(Promoter)作为基因表达的重要调控元件,对基因的表达水平有着决定性的作用。 真核生物启动子结构的复杂性是基因表达多样性的根本[1]。目前,即使是真核生物中研究最为详尽的酿酒酵母(Saccharomyces cerevisiae),预测和微调启动子的活性仍然非常困难[2-4]。 就目前而言,真核生物的代谢工程改造仍然依赖于少量定义明确的内源启动子,如GAL1-10,TEF 和LEU2 启动子等[5-7]。
热带假丝酵母(Candida tropicalis)作为一种二倍体的非常规酵母, 胞内存在一些特殊的代谢途径,能够利用木糖、烷烃和脂肪酸等一些非常规底物作为唯一碳源进行生长,并且在代谢过程中积累一些重要的化工原料,如长链二元有机酸等[8]。 因而,热带假丝酵母具有较高的工业应用前景,但是目前可供用作代谢改造的、功能确切的启动子只有少数,如3-磷酸甘油醛脱氢酶基因(GAPDH)[9]启动子和异柠檬酸裂解酶(ICL)启动子[10]。前者为转录活性较强的组成型启动子,后者为受乙酸盐强烈诱导的诱导型启动子。 在微生物代谢途径中,选用不合适的启动子有可能导致代谢途径被限速或中间产物的过量积累,而使用活性适当的启动子可以平衡微生物体内的代谢途径,降低中间产物积累,进而提高热带假丝酵母发酵过程中目的产物的产量和生产效率[11]。因此启动子数量的限制,一定程度上限制了热带假丝酵母代谢工程的研究。
作者以磷酸甘油酸激酶基因(PGK1)启动子为研究对象。 根据NCBI 数据库中已公布的PGK1 序列, 通过多重序列比对获得启动子上游的保守序列,根据保守序列设计引物PCR 扩增热带假丝酵母的PGK1 启动子, 然后对该启动子的序列进行分析并以yeGFP3 作为报告基因,通过启动子5' 端逐步截短,对启动子上游调控元件的功能进行初步鉴定和分析。
1材料与方法
1.1 材料与试剂
本文中用到的质粒Ts-CAT2-gda324-URA3和Ts-yeGFP3[12],菌株热带假丝酵母ATCC 20336、热带假丝酵母XZX 和大肠杆菌JM109 均由本研究中心保藏。 所涉及到的菌株如表1 所示:

表1 本实验所用到的菌株Table 1 Strains used in this paper
实验过程中所用酶类、载体pMD 19-T Simple、实时荧光定量所用到的试剂盒Yeast RNAiso Kit、PrimeScriptRT Reagent Kit with gDNA Eraser 和SYBRPremix Ex TaqTMKit, 均购买自Takara 公司。 质粒小提试剂盒、PCR 产物清洗试剂盒和DNA凝胶回收试剂盒,购自AxyPrep 公司。
1.2 培养基
LB 培养基:酵母粉5 g/L,蛋白胨10 g/L,氯化钠10 g/L;
MM 培养基:YNB 6.7 g/L,硫酸铵10 g/L,葡萄糖20 g/L;
SM 培养基:MM 培养基中添加0.006 g/dL的尿嘧啶;
氨苄青霉素质量浓度:100 mg/L。
1.3 方法
1.3.1 热带假丝酵母基因组DNA 的提取 热带假丝酵母基因组DNA 提取方法参考文献[12]进行操作。
1.3.2PGK1启动子的扩增与yeGFP3表达盒的构建PGK1基因的PCR 扩增: 以热带假丝酵母ATCC 20336 基 因 组DNA 为 模 板,PGKP/PGKT 为 引 物,PCR 扩增获得PGK1基因(P-ORF-T)。 经AxyPrep公司的PCR 产物清洗试剂盒纯化后, 连接到pMD 19-T Simple 载体,得到重组质粒Ts-PGK1。 重组质粒送苏州泓迅生物技术有限公司进行测序, 得到PGK1的基因序列。
以热带假丝酵母ATCC 20336 基因组DNA 为模板,PGK1/PGK3 和PGK6/PGK8 为引物,分别扩增PGK1基因的启动子PGK1P(以下称为P846)和终止子PGK1T(以下称为T)。以质粒Ts-yeGFP3为模板,PGK4/PGK5 为引物,PCR 扩增报告基因yeGFP3。
将扩增所得的P846、yeGFP3和T片段等摩尔加入融合PCR(SOE PCR)反应体系:PrimeSTARHS酶0.5 μL,5×PrimeSTARBuffer 10 μL,dNTPs 4 μL,P8461 μL,yeGFP32 μL,T1 μL 和ddH2O 31.5 μL。反应条件:98 ℃3 min;98 ℃10 s,55 ℃2 min,72 ℃2 min,10 个循环;72 ℃10 min。 以10 μL 上述融合反应液为模板,重新配置PCR 反应体系:PrimeSTARHS酶0.5 μL,5×PrimeSTARBuffer 10 μL,dNTPs 4 μL,融合反应液10 μL, 引物PGK2/PGK7 各1 μL 和ddH2O 22.5 μL。 反应条件:98 ℃3 min;98 ℃10 s,55 ℃1 min,72 ℃2 min,30 个循环;72 ℃,10 min, 获得P846-yeGFP3-T表达盒。 本实验中所用引物见表2。
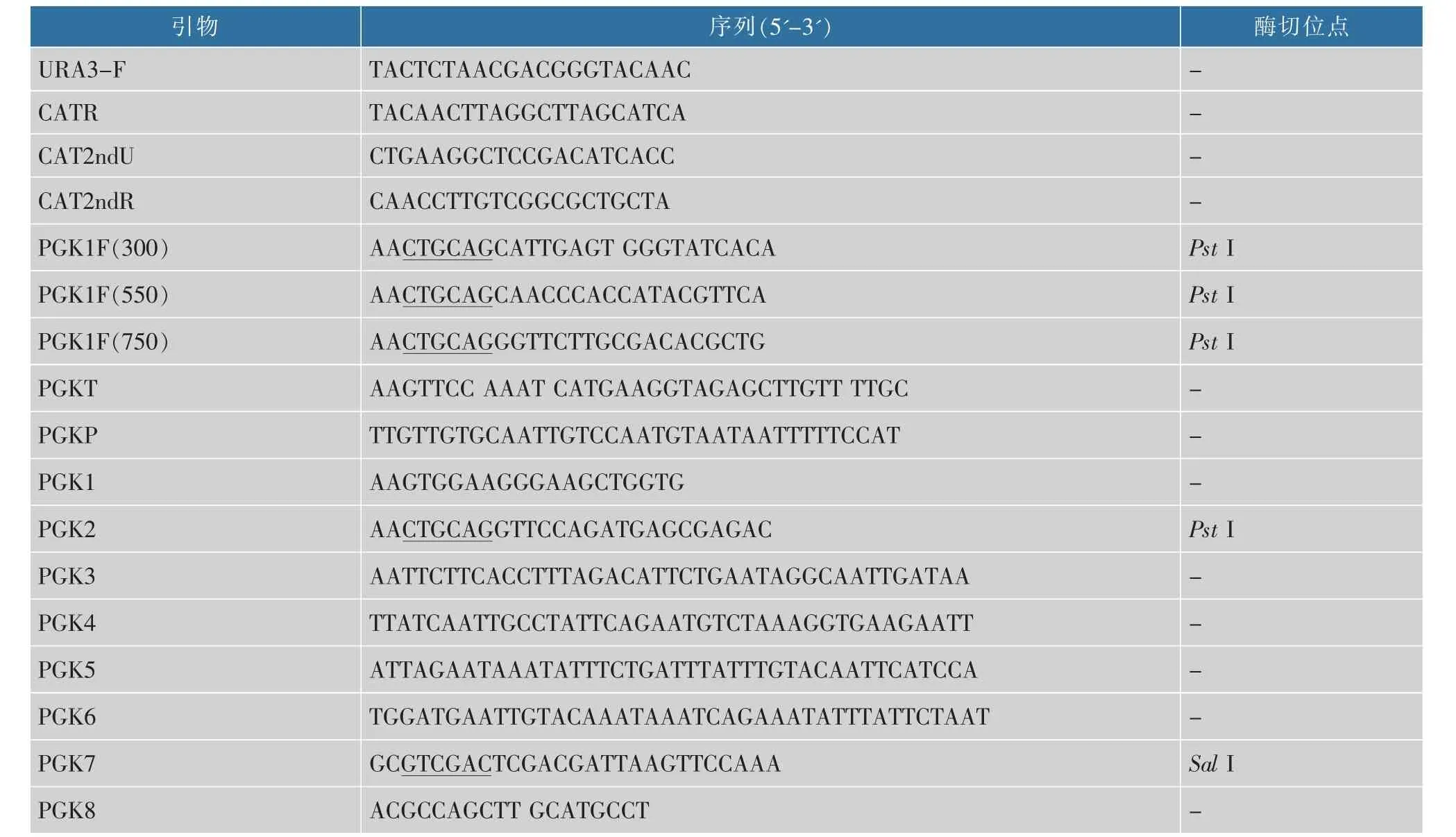
表2 本文中所用引物Table 2 Primers used in this paper
1.3.3 重组质粒Ts-CAT2-P846-yeGFP3-T-URA3的构建与转化PstI/SalI 双酶切P846-yeGFP3-T表达盒, 然后插入到经同样内切酶酶切的载体Ts-CAT2-gda324-URA3中,替换其中的gda324片段。之后将连接产物转化JM109, 获得重组质粒Ts-CAT2-P846-yeGFP3-T-URA3,并通过PstI 和SalI对重组质粒进行酶切验证。 然后以质粒Ts-CAT2-P846-yeGFP3-T-URA3为模板,CAT2ndU/CAT2ndR为引物,PCR 扩增yeGFP3基因整合表达盒CAT2-P846-yeGFP3-T-URA3-CAT2,经PCR 产物清洗试剂盒纯化后通过氯化锂转化法[13]转化尿嘧啶营养缺陷型热带假丝酵母宿主XZX。 利用2 段CAT基因片段作为同源臂, 通过同源重组将yeGFP3表达盒整合到CAT位点,经MM 平板筛得转化子后提取转化子基因组,以URA3F/CATR 为鉴定引物进行PCR验证(图1)。

图1 yeGFP3 基因表达盒的转化Fig. 1 Physical map of the expression cassette and integration of the cassette into the CAT locus of C. tropicalis XZX
1.3.4yeGFP3的检测 绿色荧光蛋白在紫外激发光(488 nm)的照射下可发射绿色荧光[14],根据这一特征对yeGFP3的表达效果进行检测。 将转化子接种到SM 培养基中,200 r/min,30 ℃培养至对数期(OD600:2~4), 然后置于4 ℃静置数小时用于稳定绿色荧光蛋白的折叠状态。 随后取1 mL 培养液12000 r/min 离心1 min 收集菌体, 经磷酸盐缓冲液(PBS)清洗菌体2 次后重悬于PBS 缓冲液中[15]。通过Nikon 80i 正置荧光显微镜以488 nm 的激发光激发后进行荧光观察并拍照记录。
将转化子培养液用PBS 缓冲液重悬, 控制OD600在0.3~0.5, 吸取200 μL 样品至96 孔黑色酶标板中, 将酶标板置于BioTek Cytation 5 细胞成像微孔板检测系统, 在488 nm/520 nm 处检测荧光强度,并对转化子的荧光信号作定量分析。 以出发菌株XZX 作为阴性对照。
1.3.5 mRNA 提取和实时荧光定量PCR 根据参考文献[12]对热带假丝酵母转化子进行实时荧光定量PCR(RT-qPCR)分析。 挑取平板上的热带假丝酵母单菌落接种于液体SM 培养基中,30 ℃、200 r/min 培养, 然后以此作为种子液转接到新鲜的SM 液体培养基中,待菌体OD600值达到1.5~2.5 后,使用酵母Total RNA 提取试剂盒提取菌株中总RNA。 使用PrimeScriptRT Reagent Kit with gDNA Eraser 试剂盒对上述RNA 进行反转录,获得单链cDNA。 使用SYBRPremix Ex TaqTM试剂盒, 以cDNA 作为模板,ACT1 为内参基因,应用CFX96 Real-Time PCR Detection System 扩增仪进行RT-qPCR 检测。 RTqPCR 反应按照95 ℃30 s;95 ℃5 s,60 ℃30 s,40个循环进行;溶解曲线按照65 ℃至95 ℃,每5 s 升温0.5 ℃进行绘制。
2结果与讨论
2.1 PGK1 基因的克隆

图25株假丝酵母PGK1 基因多重序列比对Fig. 2 Multiple sequence alignment of five PGK1 genes
参照GenBank 中已公布的5 株假丝酵母属菌株中的PGK1基因序列(登录号:FM992693.1、CP017628.1、LT635760.1、HE605202.1 和HE681724.1),分别截取5 株菌的PGK1结构基因上游约1200 bp 序列进行多重序列比对,经过比对获得一段同源性极高的序列(图2(a)),在此基础上设计PGK1基因上游引物PGKF。 以同样方法设计终止子下游引物PGKR(图2(b))。
以热带假丝酵母ATCC 20336 基因组DNA 为模板,PGKF/PGKR 为引物,PCR 扩增获得一段长度为3000 bp 的片段(图3),将片段连接到pMD 19-T Simple 载体中,然后进行测序分析。 对上述基因的测序结果进行分析, 发现该基因的编码区与Candida albicansSC5314 和Candida dubliniensisCD36 菌株的碱基序列相似度为81.8%,氨基酸相似度为92%;与Candida parapsilosisCDC317 和Candida orthopsilosisCo 90-125 菌株的碱基序列相似度为78%,氨基酸相似度为91%;与Candida intermediaCBS 141442 菌株的碱基序列相似度为76%,氨基酸相似度为86.8%。 因此可以判断所扩增得到的基因为PGK1。
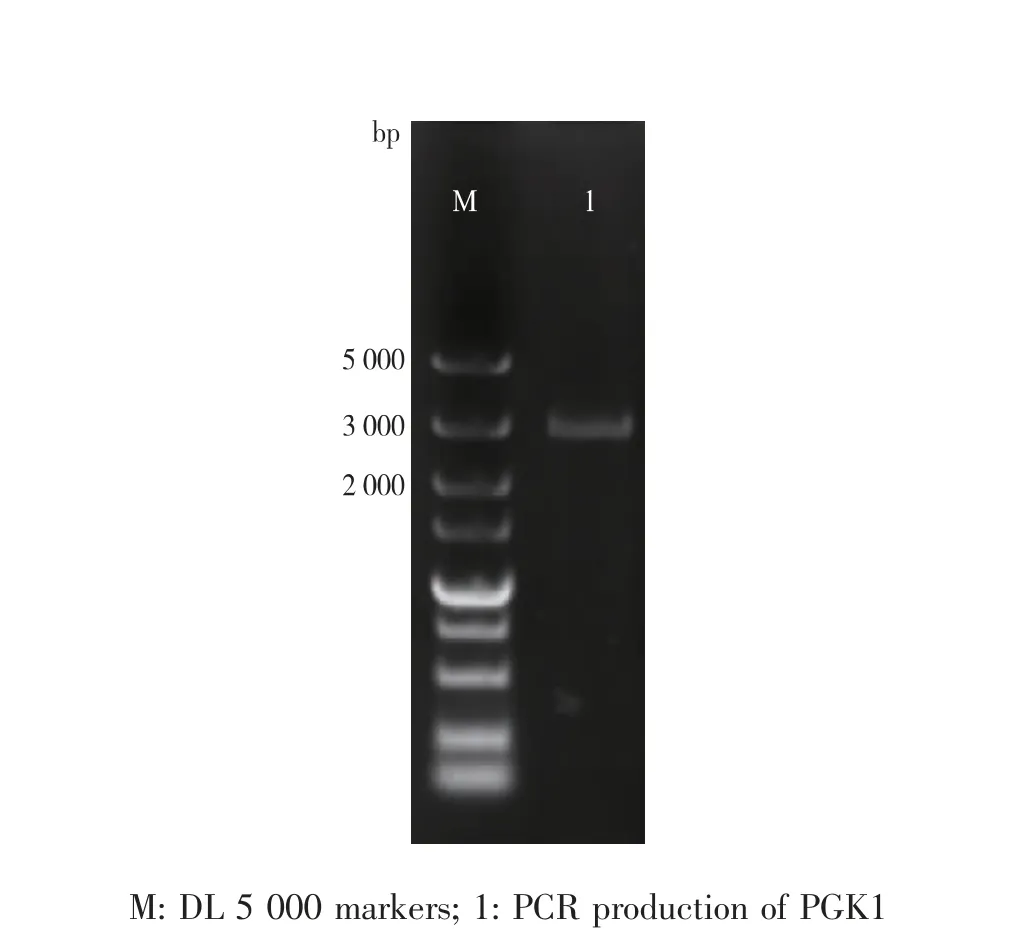
图3 PGK1 基因克隆与电泳分析Fig. 3 Amplification of PGK1 gene
2.2 PGK1 启动子的功能鉴定
重组质粒Ts-CAT2-P-yeGFP3-T-URA3构建过程如图4(c)所示。 分别PCR 扩增报告基因yeGFP3、PGK1的启动子P846和终止子T,通过融合PCR 构建yeGFP3表达盒P846-yeGFP3-T(图4(a))。 用表达盒替换载体Ts-CAT2-gda324-URA3中的gda324片段, 获得重组质粒Ts-CAT2-P846-yeGFP3-TURA3。 用PstI 和SalI 对重组质粒进行双酶切,得到5 kb 的载体片段和2.1 kb 的yeGFP3表达盒片段(图4(b))。将构建完成的表达盒通过氯化锂转化法转化XZX 菌株并筛选获得转化子,阳性转化子命名为C. tropicalisP-4(图6,泳道4)。 在荧光显微镜下观察到P-4 菌株具有清晰的绿色荧光(图7),由此可以确认PGK1启动子具有活性。
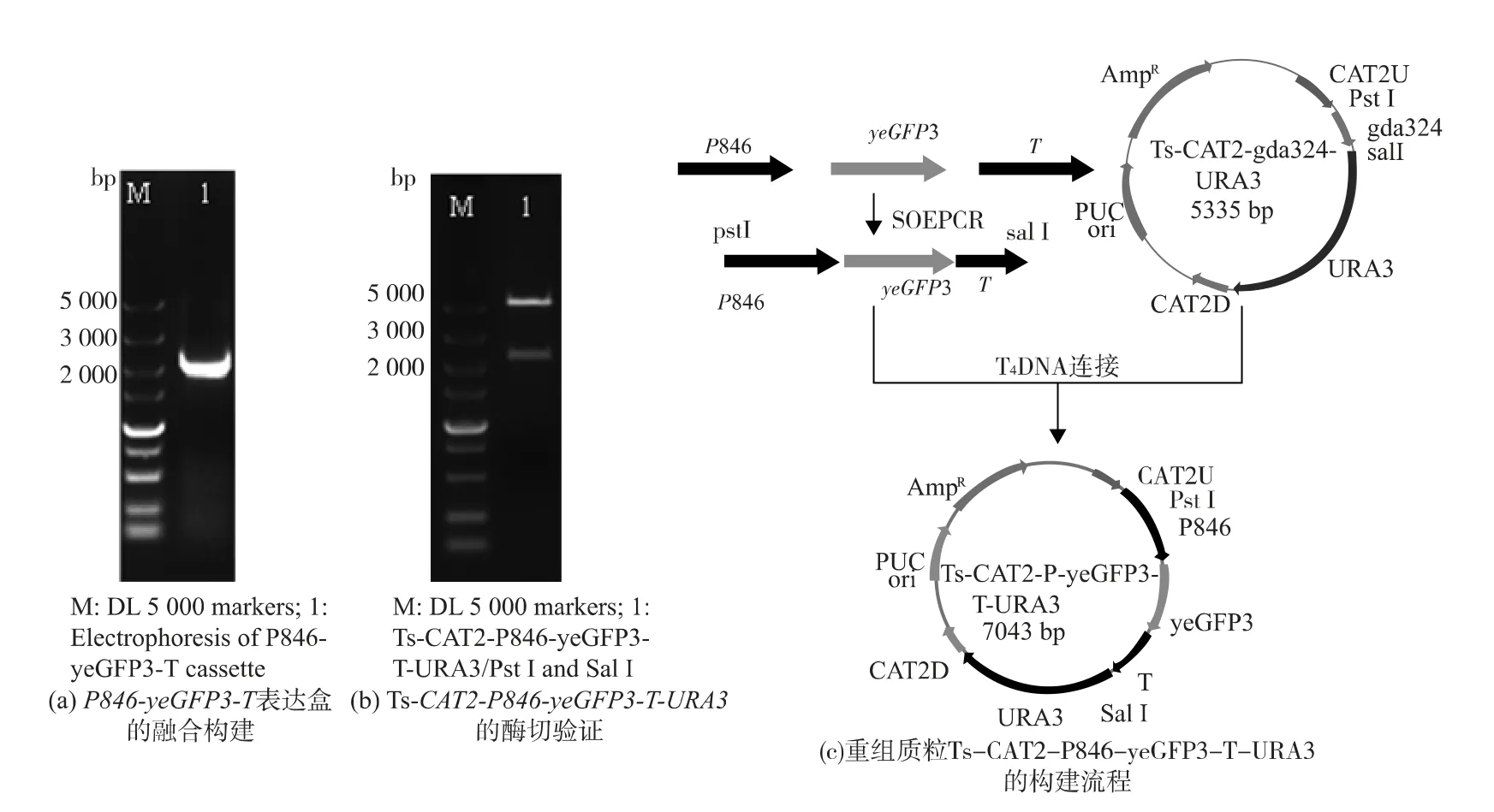
图4 P846-yeGFP3-T 表达盒的融合构建、Ts-CAT2-P846-yeGFP3-T-URA3 的酶切验证、 重组质粒Ts-CAT2-P846-yeGFP3-T-URA3 的构建流程Fig. 4 Construction of P846-yeGFP3-T cassette by SOE PCR,Restriction analysis of plasmid Ts-CAT2-P846-yeGFP3-TURA3 and construction of the recombinant plasmid Ts-CAT2-P846-yeGFP3-T-URA3
2.3 启动子的截短与yeGFP3 表达盒构建
通过在线启动子分析网站promoter 2.0(http://www.cbs.dtu.dk/services/Promoter/) 分析PGK1启动子序列, 分析结果显示在启动子-500 bp 处存在一个可能的功能元件, 在-139 bp 处存在一个可能的TATA 框,-84 bp 处和-240 bp 处存在2 个可能的CAAT 框。因此将PGK1启动子划分为300 bp(以下称该长度启动子为P300)、550 bp(以下称该长度启动子为P550) 和750 bp (以下称该长度启动子为P750)3 个长度进行分析。
分别以PGK1F(300)/PGK7、PGK1F(550)/PGK7和PGK1F(750)/PGK7 为引物,重组质粒Ts-CAT2-P846-yeGFP3-T-URA3为模板,PCR 扩增得到不同长度PGK1启动子调控下的yeGFP3表达盒,用PstI/SalI 双酶切后连接到经同样内切酶酶切的载体Ts-CAT2-gda324-URA3中, 分别构建重组质粒Ts-CAT2-P300-yeGFP3-T-URA3、Ts-CAT2-P550-yeGFP3-T-URA3和Ts-CAT2-P750-yeGFP3-TURA3。 用PstI/NdeI 双酶切, 得到启动子加部分yeGFP3的片段,大小依次为530、780、980 bp 和1076 bp,和2 段约3000 bp 的片段(因大小相差过小电泳难以分开,图5)。 分别以上述3 个重组质粒为模板,CAT2ndU/CAT2ndR 为引物, 扩增yeGFP3表达盒CAT2 -P300 -yeGFP3 -T -URA3 -CAT2、CAT2-P550-yeGFP3-T-URA3-CAT2和CAT2-P750-yeGFP3-T-URA3-CAT2。
2.4 yeGFP3 基因表达盒的转化与鉴定
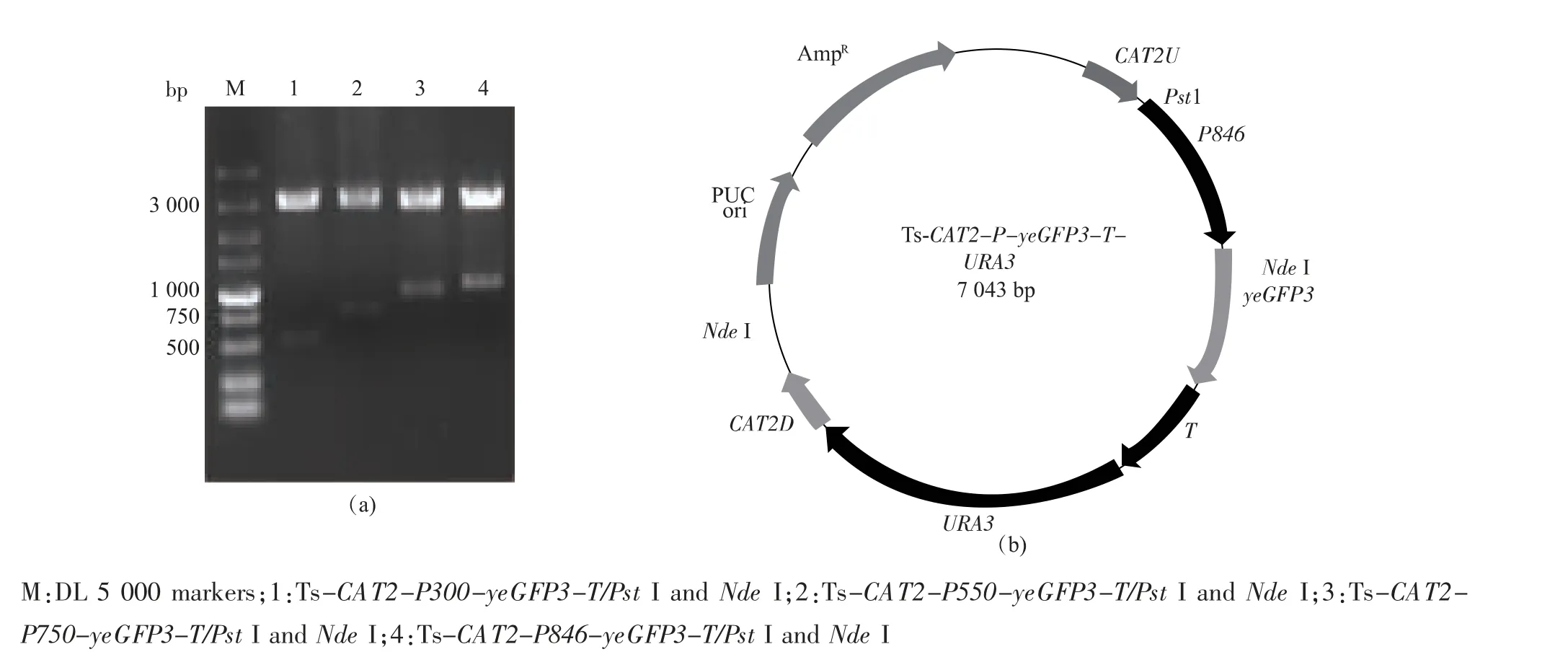
图5 重组质粒的酶切验证Fig. 5 Restriction analysis of recombinant plasmids
分别将4 个不同长度启动子调控下的yeGFP3表达盒转化XZX 菌株, 用MM 平板筛选转化子,提取转化子基因组,使用URA3F/CATR 引物进行PCR验证, 阳性转化子可扩增得到2.3 kb 的条带,而XZX 菌株则无特异性扩增条带(图6)。 分别命名阳性转化子为P-1、P-2、P-3 和P-4。
2.5 转录水平和翻译水平表征各截短启动子的强度
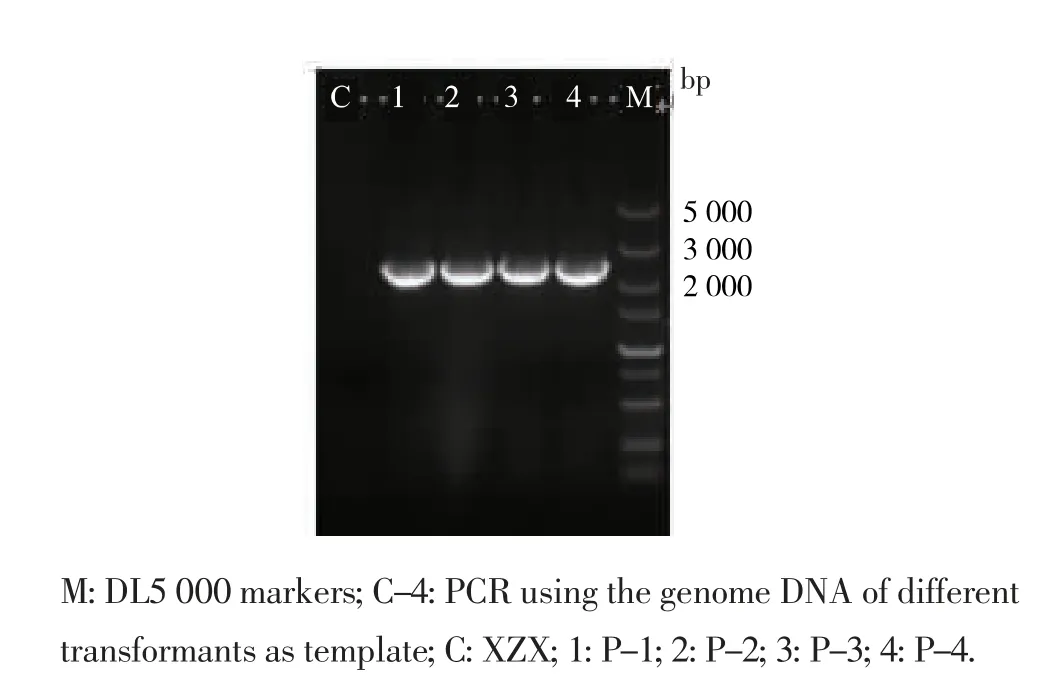
图6 截短启动子的yeGFP3 表达盒整合转化子的PCR 鉴定Fig.6 Identification of truncated promoter cassette by PCR
PGK1启动子为组成型启动子, 能够在菌体生长过程中稳定的进行表达,且不需要添加诱导物。在SM 液体培养基中分别培养P-1、P-2、P-3、P-4 和出发菌株XZX 至OD6002~4, 然后用荧光显微镜观察记录各转化子中yeGFP3基因的表达情况并进行定性分析。荧光显微镜结果显示,yeGFP3在P-1 和P-2 菌株中表达极弱,几乎无法观察到荧光;P-3 菌株中yeGFP3表达水平较高, 在紫外激发光下可以看见明显的绿色荧光; 而在P-4 中yeGFP3表达水平最高,能够清晰的观察到强烈的绿色荧光(图7)。荧光检测结果显示,PGK1启动子序列越完整,细胞的荧光强度越强。
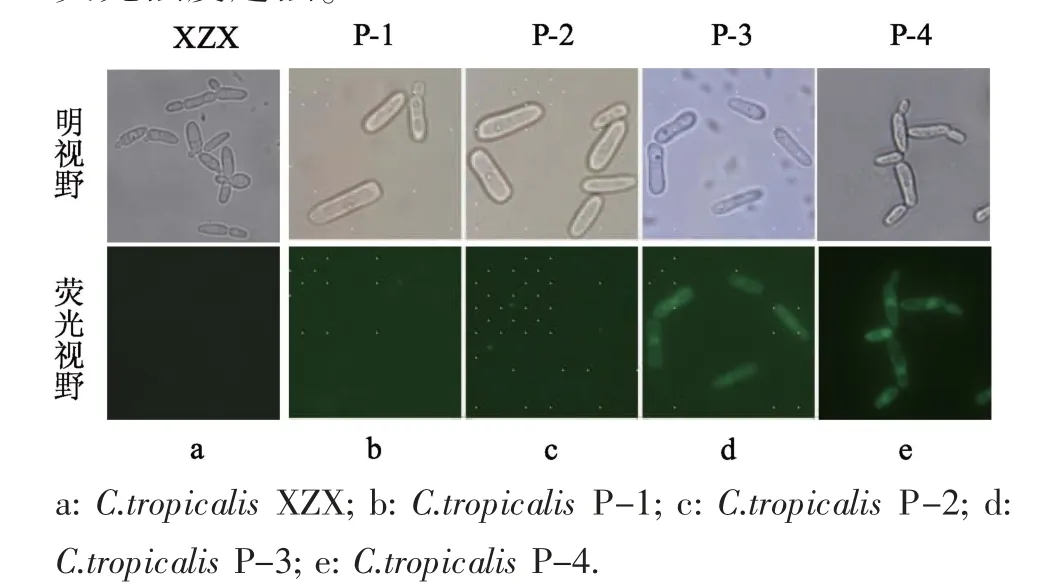
图7 热带假丝酵母转化子中绿色荧光蛋白的检测Fig. 7 Observation of yeGFP3 in C. tropicalis XZX
使用BioTek Cytation 5 细胞成像微孔板检测系统对4 个转化子中绿色荧光蛋白的表达水平进行定量分析, 检测结果与荧光显微镜观察结果一致,P-1、P-2 和P-3 菌株中的相对荧光强度较P-4 菌株低,分别为P-4 菌株的4.98%、9.84%和48.4%(图8)。
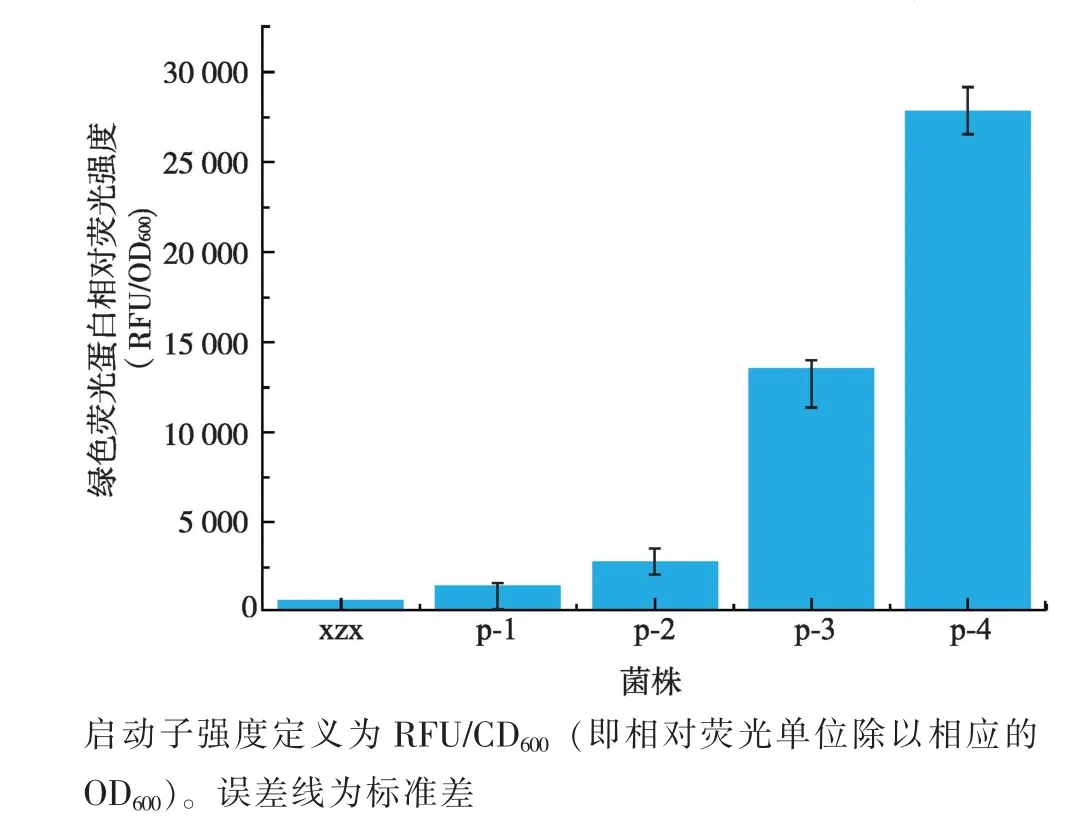
图8 热带假丝酵母转化子的相对荧光强度(RFU/OD600)Fig. 8 Relative fluorecence intensity of the five strains
由于转化子P-1 和P-2 所表达的绿色荧光极为微弱,为进一步确定550 bp 以下的启动子片段是否具有转录活性,并分析不同长度启动子调控下的蛋白表达水平与mRNA 转录水平是否一致,分别提取上述5 株菌的总RNA, 并进行RT-qPCR 分析。RT-qPCR 实验结果显示P-1 和P-2 菌株中PGK1启动子仅有较低的转录活性, 转化子中yeGFP3的转录水平分别为P-4 菌株的5.6%、13.8%,而yeGFP3在P-3 菌株中的转录水平达到了P-4 菌株的60%(图9)。 该数据与BioTek Cytation 5 细胞成像微孔板检测系统结果一致。
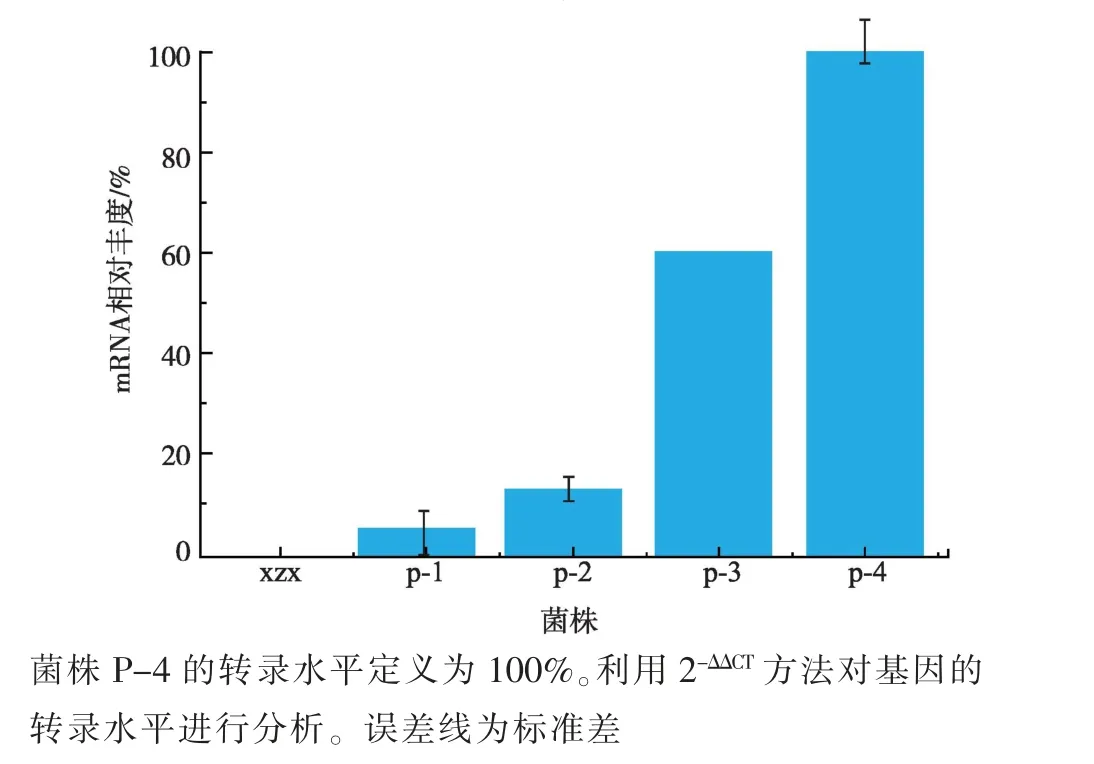
图9 不同热带假丝酵母转化子中yeGFP3 基因的mRNA相对丰度分析Fig.9 mRNA levels of different yeGFP3 genes in transformants
3结 语
GFP 作为一种从水母中克隆得到的荧光蛋白,无需辅助因子,经紫外光激发即可发射绿色荧光[16],被广泛用于启动子的功能研究。 本文选用密码子优化的酵母增强型GFP(yeGFP3)作为报告基因[17],对热带假丝酵母的PGK1启动子功能进行鉴定。
PGK1 是糖酵解途径中的关键酶,在糖酵解第2个阶段的第2 步中催化1,3-二磷酸甘油酸转变3-磷酸甘油酸并产生1 分子ATP。PGK1启动子在酵母中具有较强的启动活性,在酵母代谢改造中常被用作强启动子启动外源基因的转录[18]。 且PGK1启动子是组成型启动子,可以在廉价碳源如葡萄糖中稳定表达外源蛋白,不需要添加诱导物,因此选择热带假丝酵母中的PGK1启动子为研究对象。 根据在线启动子分析网站(http://www.cbs.dtu.dk/services/Promoter/)对PCK1启动子调控元件的预测结果,以XZX 为出发菌株, 构建了P-1、P-2、P-3 和P-4 四株重组菌,分别以不同长度(300、550、750 bp 和846 bp)的PGK1启动子来调控yeGFP3表达。 通过比较4 株重组菌中yeGFP3的表达差异来探索PGK1启动子的调控元件所在区域。 实验结果显示:550 bp以下的PGK1启动子片段转录活性极低且重组菌的荧光微不可查, 推测在-550 bp 以下无功能元件或者在-550 bp 处功能元件被截断, 以至于未能发挥应有的辅助转录功能;750 bp 的PGK1启动子片段有较明显的转录活性且重组菌的荧光清晰可见,该结果可证明-750 bp 之前确有功能元件且为UAS,但具体序列大小与存在位置需后续实验做进一步的确认;最后846 bp 的PGK1启动子片段具有高于750 bp 的转录活性且重组菌荧光信号稳定,可能在750 bp 上游至少还有1 个UAS。 因此初步判断PGK1启动子中至少存在2 个UAS。
外源基因的转录是一个较为复杂的过程, 基于分离不同长度的启动子功能的比较研究, 下一步的工作是对PGK1启动子的2 个可能的UAS 进行详细分析,分离获得明确的功能序列。 再对PGK1启动子进行改造, 将获得的UAS 分别在PGK1上游进行串联表达,得到一个转录活性更强的杂合启动子,为以后在热带假丝酵母中高效表达外源基因奠定基础。
猜你喜欢
昆明医科大学学报(2022年1期)2022-02-28
海南医学(2021年9期)2021-05-26
女报(2020年7期)2020-08-17
中国保健营养(2019年3期)2019-10-21
儿童故事画报(2019年8期)2019-08-14
艺术启蒙(2018年8期)2018-08-22
名人传记·财富人物(2017年9期)2017-11-02
名人传记·财富人物(2017年9期)2017-11-02
Coco薇(2016年8期)2016-10-09
小火炬·智漫悦读(2015年4期)2015-06-05
