
Au7团簇吸附O2和CO及其对CO的催化反应机理研究
2020-05-15 10:59邓开明
原子与分子物理学报 2020年3期
陈 宣, 邓开明
(1.南京信息工程大学 物理与光电工程学院, 南京 210044; 2.南京理工大学 理学院, 南京 210094)
1 前 言
金历来被认为比其它金属的活性要低的多,其表面很难吸附大量的气体分子,不具有催化特性[1-3].自Haruta等人发现纳米金颗粒有着活跃的催化性质[4]以来,金团簇对CO催化氧化反应成为一个十分活跃的研究领域[5-10].大量的实验[11-13]和理论[14, 15]研究表明,当金颗粒尺寸为2~5nm时具有良好的催化活性,反应的关键过程是氧分子在团簇上的活化,形成一种超氧状态[O2]-,然后同吸附的CO分子进行反应.然而,金纳米颗粒能够很好的吸附CO分子,但很难牢牢的吸附O2分子[16].实验和理论研究表明当Aun团簇有未配对电子时才能对O2分子展现出良好的吸附能力[13].
目前,Au团簇的CO催化氧化反应机理也引起了大量的理论研究[17-23].在Au10团簇的研究中发现,O2分子在Au10团簇上有可能分裂为两个O原子,从而降低CO氧化反应时的反应势垒[21].在硅烯表面上Au原子对C催化氧化的研究中表明,CO先吸附在Au上然后在和O2分子反应,在反应过程中形成OCOO结构的中间态[22].Lin等人在研究Au2n-(n=1-4)团簇CO催化氧化反应时提出,CO和O2在反应过程中间有可能形成一种稳定的CO3结构[23].而Wang等人对Au3-,Au3和Au3+团簇的理论研究表明,离子态会影响CO催化氧化活性,在反应过程中CO不能直接与吸附在金团簇上的O2分子反应,而时在反应过程中形成了OCOO结构的中间态,并指出反应过程中并不一定会形成稳定的CO3结构[24].
Wang等人在研究了Au7团簇在TiO2(110)面上的CO催化氧化反应时,主要研究了三维立体结构的Au7团簇对CO催化氧化反应机理以及负载在反应中起的作用[25].最近,Gruene等人在实验中发现了二维Cs平面结构的Au7团簇[26],其较小的尺寸在无负载情况下也有可能成为CO氧化的催化剂.本文采用密度泛函理论的方法,研究了Au7团簇吸附O2、CO分子情况,并讨论了CO在Au7团簇上催化氧化的反应机理.
2 计算方法
本文采用相对论密度泛函理论中的广义梯度近似(GGA)[27,28],Perdew,Burke和Ernzerhof交换关联修正函数[29]和极化函数扩展的双数值原子轨道DND基组,也就是说函数中包含高于自由原子中的最高占据轨道角动量一级的角动量.计算中采用自旋非限制近似求解Kohn-Sham[30]自洽场方程.结构优化采用了Broyden-Fletcher-Goldfarb-Shanno[31]方法,在没有任何参数限制(如对称性,键长,键角)条件下,以梯度变化小于10-3a.u、位移变化小于10-3a.u.和能量变化小于10-5a.u.作为收敛标准,自洽过程是在能量和电子密度的收敛标准为10-6a.u.下完成.过渡态的搜索采用的是线性同步度越(linear synchronous transit, LST)和四极同步度越(quadratic synchronous transit, QST)方法.
3 结果与分析
图1给出了Au7团簇两种优化的几何结构,分别是对称性Cs的平面结构[25]和对称性D5h的立体结构.表1列出了Au7团簇两种结构的结合能(Eb)和其吸附一个CO分子或一个O2分子时的吸附能(Eads).Eb是组成分子的各原子能量之和减去分子总能量,它反映了各同分异构体的热力学稳定性.Eads是Au7团簇能量与O2(或CO)分子能量之和减去Au7-O2(或Au7-CO)团簇的能量.由表1可知,Cs结构的Au7团簇结合能比D5h结构的结合能大,说明C2v结构较稳定,Au7团簇更倾向与平面结构.此外,Cs结构的Au7团簇的Eads(O2)和Eads(CO)也比D5h结构的Au7团簇大,表明其对O2和CO的吸附能力较强.因此,以下仅讨论Cs结构的Au7团簇吸附O2、CO分子情况以及其CO催化氧化的反应机理.

图1 Au7团簇的几何优化结构:(a)CS对称性结构,(b)D5h对称性结构.Fig.1 Optimized geometries of Au7: (a) CS symmetric structure, (b) D5h symmetric structure.
表1 Au7团簇的结合能以及其CO和O2的吸附能
Table 1 The binding energies of Au7clusters as well as the adsorption energies of CO and O2on Au7

CsD5hEb /eV11.7211.18Eads(O2)/eV0.640.58Eads(CO)/eV1.261.16
3.1 Au7团簇对O2分子的吸附性质
O2分子在Au7团簇上的吸附位置可以分为三类,分别为吸附在Au原子上,吸附在Au-Au键上,和吸附在Au原子形成的面上.计算结果表明O2分子更容易吸附在Au原子上.图2 给出了Au7团簇吸附一个O2分子时较稳定的三种几何结构.表2列出了它们的结合能、吸附能及其相关键长.由结合能可以发现,Au7团簇吸附一个O2分子时,最稳定的吸附位置为O2分子吸附在Au1原子上,如图2(a)所示,而O2分子吸附在Au7、Au5原子上时[图2(b)和(c)],其结合能比最稳定结构的结合能小0.09 eV和0.18 eV.此外,在这三种吸附位置中,O-O键长都有所增长,从O2分子的0.123 nm增长到0.126 nm左右;而Au-O键长随着结合能和吸附能的减少而变长.图3给出了Au7团簇的电子最高占据轨道图.从该图可以看出,电子主要分布在Au1、Au5和Au7原子周围,说明与其它位置相比,当O2分子吸附在这三个位置时,O2分子更容易得到电子,形成超氧状态.

图2 Au7O2分子的几何结构图:(a)O2分子吸附在Au1原子位置,(b)O2分子吸附在Au7原子位置,(c)O2分子吸附在Au5原子位置.Fig.2 Optimized geometries for the complexes of Au7O2: (a) O2 is adsorbed on Au1 atom, (b) O2 is adsorbed on Au7 atom, (c) O2 is adsorbed on Au5 atom.
表2 O2分子分别吸附在Au1、Au7、Au5上时的结合能、吸附能及其相关键长
Table 2 The binding energies, the adsorption energies and the bond lengths for the structures of O2adsorbed on Au1, Au7 and Au5 atoms, respectively.

O2-Au1O2-Au7O2-Au5d(Au-O)/nm0.2130.2150.217d(O-O)/nm0.1260.1270.126Eb /eV19.0518.9618.87Eads/eV0.640.550.46

图3 Au7团簇电子最高占据轨道图.Fig.3 Typically contour plots of HOMO of Au7 cluster
表3给出了Au7团簇吸附多个O2分子的平均吸附能及相关键长.由表3可以看出,随着O2分子吸附数目的增加,O-O键略有减小,但Au-O键明显增长.平均吸附能也随着吸附O2数目的增加而明显降低,并且减小的幅度很大.因此Au7团簇吸附多个O2分子的可能性不大.
表3 多个O2分子吸附在Au7团簇表面上时,各稳定结构的平均键长及平均吸附能
Table 3 The average adsorption energies and the average geometrical parameters for multiple O2adsorption on the Au7cluster
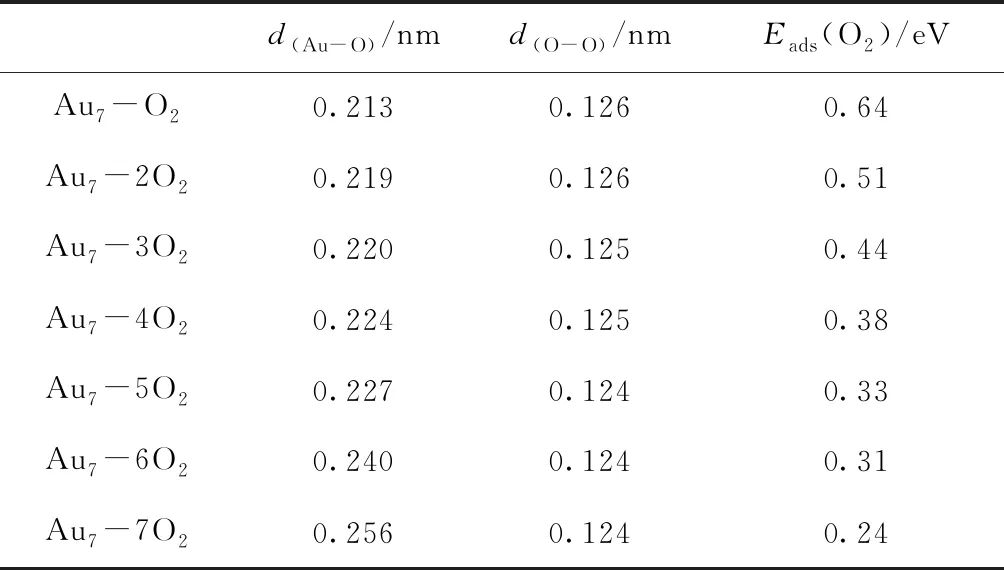
d(Au-O)/nmd(O-O)/nmEads(O2)/eVAu7-O20.2130.1260.64Au7-2O20.2190.1260.51Au7-3O20.2200.1250.44Au7-4O20.2240.1250.38Au7-5O20.2270.1240.33Au7-6O20.2400.1240.31Au7-7O20.2560.1240.24
3.2 Au7团簇对CO分子的吸附性质
图4给出了Au7团簇吸附一个CO时四种较稳定的几何优化结构.与吸附O2分子不同,Au7团簇吸附CO分子时,Au7团簇的结构发生了很大的扭曲.表4给出了这四种结构的结合能、吸附能和相关的键长.当CO分子吸附在Au6原子、Au7原子或Au6-Au7键上时,Au7团簇的结构发生扭曲,形成最稳定的吸附结构[图4(a)],其吸附能为1.26 eV.此外,当CO分子吸附在Au1、Au3和Au5原子位置时,也可以得到较为稳定的结构,其结合能比最稳定结构的结合能小0.16、0.11和0.19 eV.与O2分子的吸附能相比,CO分子的吸附能要大于对O2分子的吸附能,这说明Au7团簇更容易吸附CO分子.

图4 Au7团簇吸附一个CO分子的几何结构图:(a)CO分子吸附在Au6原子位置,(b)CO分子吸附在Au1原子位置,(c)CO分子吸附在Au3原子位置,(d)CO分子吸附在Au5原子位置.Fig.4 Optimized geometries for the complexes of Au7CO: (a) CO is adsorbed on Au6 atom, (b) CO is adsorbed on Au1 atom, (c) CO is adsorbed on Au3 atom, (d) CO is adsorbed on Au5 atom.
表4 CO分子分别吸附在Au1、Au3、Au5和Au6原子时的结合能、吸附能及其相关键长
Table 4 The binding energies, the adsorption energies and the bond lengths for the structures of CO adsorbed on Au1, Au3, Au5 and Au6 atoms, respectively.
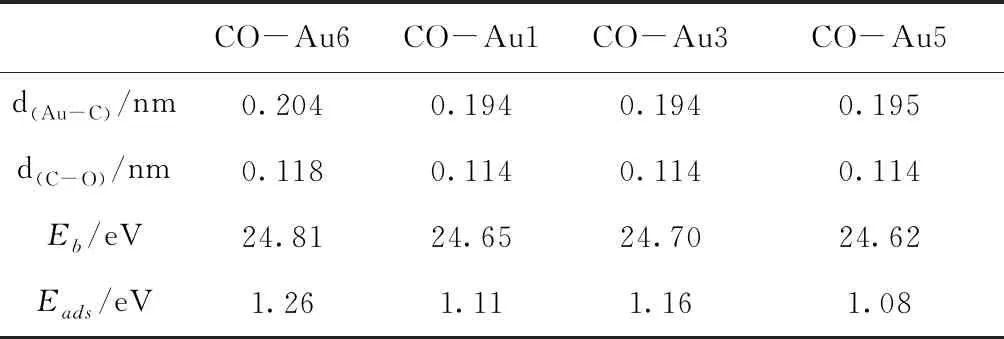
CO-Au6CO-Au1CO-Au3CO-Au5d(Au-C)/nm0.2040.1940.1940.195d(C-O)/nm0.1180.1140.1140.114Eb/eV24.8124.6524.7024.62Eads/eV1.261.111.161.08
表5给出了Au7团簇吸附多个CO分子时的平均吸附能以及平均键长.由表5可以看出,虽然平均吸附能随着吸附CO分子个数的增多而有所减小,但减小的幅度不是很大,这表明Au7团簇有可能同时吸附多个CO分子.此外,除了吸附一个CO分子的情况外,Au7团簇吸附多个CO分子时,Au-C键长及C-O键长均变化不大.
表5 多个CO分子吸附在Au7团簇表面上时,各种稳定结构的平均键长及平均吸附能
Table 5 The average adsorption energies and the average geometrical parameters for multiple CO adsorption on the Au7cluster

d(Au-C)/nmd(C-O)/nmEads(CO)/eVAu7-CO0.2030.1171.26Au7-2CO0.1940.1141.22Au7-3CO0.1940.1141.20Au7-4CO0.1950.1141.13Au7-5CO0.1980.1151.07Au7-6CO0.1960.1141.03Au7-7CO0.1970.1150.91
3.3 Au7团簇对CO催化氧化反应机理
由上文分析可知,Au7团簇更容易吸附CO分子,因此Au7团簇CO催化氧化反应过程应该是CO分子先吸附在Au7团簇上,然后和O2分子发生反应.图5给出了Au7团簇CO催化氧化过程的势能图以及各状态的几何结构图.表6列出了反应过程中各状态的几何结构参数.由图5可知,在初态IS中,以Au7-CO结构为基础,吸附一个O2分子,其吸附能为0.40 eV,形成了CO和O2分子共吸附在Au7团簇上的结构.IS克服0.18 eV的势垒,到达过渡态TS1,CO与O2相互靠近,两者距离d(O-C)从0.291 nm缩短为0.198 nm,并且O-O键开始增长.过渡态TS1态释放0.23 eV能量后,形成一种较为稳定的OCOO中间产物.在中间态MS中,O-O键长有了明显的增长,变为0.144 nm,并且CO与O2进一步靠近,d(O-C)缩短为0.133 nm.MS克服0.34 eV的势垒到达过渡态TS2.在TS2中,O-O键正在断开,其之间的距离增长为0.173 nm,CO2分子即将形成.最终,CO2分子脱离Au7团簇,释放能量3.00 eV.从整体上看,Au7团簇CO催化氧化反应需克服的最高势垒仅为0.34 eV,说明此反应是可以进行的.

图5 CO在Au7团簇上的催化氧化反应过程的势能图,以Au7团簇、O2分子和CO分子的能量和作为零势能,能量单位为eV.IS为初始状态,TS1和TS2为过渡态,MS为中间态,FS为终态 Fig.5 Energy diagrams for CO oxidation on Au7 cluster.The corresponding initial, intermediate, transition and final states are presented.The sum of energies of free Au7, O2, and CO is set to zero as a reference.All the energies are in eV.
4 结 论
本文采用密度泛函理论,研究了Au7团簇对O2和CO分子吸附的情况,探究了CO在Au7团簇上的催化氧化反应机理.研究发现:Au7团簇吸附一个O2分子的吸附能为0.64 eV,但在吸附多个O2分子时,平均吸附能有了明显的下降,表明Au7团簇进行多吸附O2分子的可能性不大.Au7团簇吸附一个CO分子的吸附能为1.26 eV,且在吸附多个CO分子时,平均吸附能减小幅度不大,说明Au7团簇有可能吸附多个CO分子.由吸附能可知,Au7团簇更倾向于吸附CO分子,并且Au7团簇的CO催化氧化反应过程为CO分子先吸附在Au7团簇上,然后和O2分子发生反应.由反应势能可知,整个反应要克服的最高势垒仅为0.34 eV,因此Au7团簇有望成为良好的CO氧化催化剂.
表6 Au7团簇CO催化氧化反应中各状态几何结构参数,其中d(C-O)为CO中C原子与O原子之间的距离;d(Au-C)为CO与Au7团簇之间的距离;d(O-C)为CO与O2之间的距离;d(O-O)为O2中两个O原子之间的距离;d(Au-O)为O2与Au7团簇之间的距离
Table 6 The geometrical parameters for initial, intermediate, and transition states, respectively.Thed(C-O)is the distance between C and O atoms in CO; the d(Au-C)is the distance between CO and Au7; the d(O-C)is the distance between CO and O2; the d(O-O)is the distance between O and O atoms in O2;the d(Au-O)is the distance between O2and Au7.
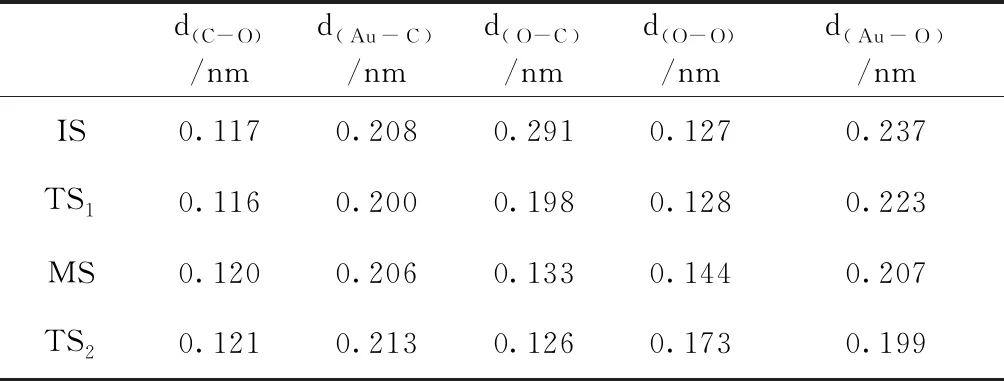
d(C-O)/nmd(Au-C)/nmd(O-C)/nmd(O-O)/nmd(Au-O)/nmIS0.1170.2080.2910.1270.237TS10.1160.2000.1980.1280.223MS0.1200.2060.1330.1440.207TS20.1210.2130.1260.1730.199
猜你喜欢
大学物理(2022年9期)2022-09-28
建材发展导向(2021年14期)2021-08-23
物理通报(2020年7期)2020-07-01
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中国煤层气(2019年2期)2019-08-27
枣庄学院学报(2015年5期)2016-01-09
中学化学(2015年8期)2015-12-29
原子与分子物理学报(2015年3期)2015-11-24
火炸药学报(2014年1期)2014-03-20
火炸药学报(2014年1期)2014-03-20
