
肉桂蒸油剩余物中活性成分的提取、纯化及其抗氧化活性研究
2020-05-12 02:39毕良武曾维星赵振东陈玉湘张笮晦
林产化学与工业 2020年2期
程 贤, 毕良武*, 曾维星, 赵振东, 陈玉湘, 张笮晦
(1.中国林业科学研究院 林产化学工业研究所;生物质化学利用国家工程实验室;国家林业和草原局林产化学工程重点实验室;江苏省生物质能源与材料重点实验室;江苏省林业资源高效加工利用协同创新中心,江苏 南京210042;2.广西中医药大学 药学院;广西中药药效研究重点实验室,广西 南宁530200;3.广西庚源香料有限责任公司,广西 东兴538100)
肉桂(Cinnamomum cassiaPresl)主要分布在我国的广西,此外,印度、老挝以及越南等地也有种植[1]。 肉桂作为一种常见的中药材,其主要活性成分为肉桂油[2]。 肉桂油常用于传统中药复方,肉桂油深加工产品可用于食品防腐剂、香料、抑菌剂、杀虫剂和功能性材料等领域[3]。 肉桂油通常由干燥的枝、叶经水蒸气蒸馏所得,具有多种药理作用。 除此以外,肉桂油提取后的剩余物中含有大量的多酚和黄酮,其中多酚类成分包括儿茶素、原儿茶酸、羟基查尔酮和原花青素等,黄酮类成分包括槲皮素、山柰酚和香豆素等[4-6]。 肉桂中的多酚和黄酮类成分具有抗菌、抗氧化、抗肿瘤等生物活性,是天然抗氧化剂的重要来源,具有广阔的应用前景[7-9]。 Prasad 等[10]对肉桂中的总酚类物质进行了体内和体外的抗肿瘤实验,结果表明:肉桂中的总酚类物质具有良好的抗肿瘤活性。 Li 等[11]研究表明肉桂中含有黄烷醇等活性多酚类抗氧化物质,具有增强胰岛素的功能。 钟益宁等[12]通过测定肉桂多酚对4 种菌的体外抗菌活性,结果发现:肉桂多酚具有较强的体外抗菌作用。 曾华君等[13]利用细胞分子生物学的手段证明肉桂黄酮成分能有效抑制6-羟基多巴胺诱导的PC12 细胞损伤(帕金森模型),其作用机制在于肉桂黄酮中含有的多个酚羟基具有较高的还原性,因此能有效抑制细胞膜脂质氧化。 林款等[14]对分离得到的肉桂黄酮进行体外抗氧化测试,研究表明:肉桂黄酮显示出较好的抗氧化效果,效果接近阳性对照Vc。 由此可见,肉桂多酚及黄酮在治疗心血管疾病、糖尿病等方面具一定的药用价值;在开发食品天然防腐抗菌剂及抗氧化剂等方面具有广阔的应用前景。 然而,肉桂提取精油后的残渣并没有得到充分的利用,对不同部位肉桂蒸油剩余物(CR)化学成分的抗氧化活性比较研究也未见报道。 因此,本研究以桂皮蒸油剩余物(BR)、桂叶蒸油剩余物(LR)、桂枝蒸油剩余物(TR)为原料,经过醇提取和大孔树脂纯化处理,测定多酚和黄酮含量,并测定其总抗氧化能力、DPPH·和·OH 的清除能力,旨在深入探索不同部位CR 的抗氧化活性,以期为肉桂资源的综合开发利用提供参考。
1 实 验
1.1 原料、试剂与仪器
干燥的肉桂叶、肉桂枝、肉桂皮,广西庚源香料有限责任公司,粉碎后过0.25 mm 筛备用。 AB-8 大孔树脂,购自河北宝恩树脂科技有限公司;芦丁(HPLC 纯度>98%),购自成都德思特生物科技有限公司;没食子酸(HPLC 纯度>98%)、福林酚试剂,购自上海源叶生物科技有限公司;总抗氧化能力(TAOC)测定试剂盒,购自南京建成生物工程研究所;无水乙醇、甲醇、亚硝酸钠、硝酸铝、氢氧化钠、碳酸钠、抗坏血酸(Vc)、1,2-二苯基-2-三硝基苯肼(DPPH)、水杨酸钠、硫酸亚铁和双氧水,均为市售分析纯。
UV-1800 紫外-可见分光光度计,岛津LC-20A 高效液相色谱仪,配有CBM-20A 系统控制器、LC-20AT 泵、SIL-20A 自动进样器、CTO-20A 柱温箱、SPD-M20A UV-VIS 检测器,日本岛津公司;LD-UPW-V超纯水制备仪;DZF-6020 真空干燥箱。
1.2 肉桂蒸油剩余物活性成分的制备
1.2.1提取取干燥的肉桂叶、肉桂枝和肉桂皮分别粉碎、过筛,转移到水蒸气蒸馏装置中,按照料液比1 ∶50(g ∶mL)加入蒸馏水,于100 ℃恒温水浴加热3 h,采用抽滤方式分离。 肉桂蒸油剩余物(CR)按照料液比1∶50(g∶mL)进行超声波乙醇提取30 min,采用抽滤方式分离。 将2 次滤液合并后减压浓缩至原有体积的1/50,2 ~8 ℃冷藏备用。 取2 mL 真空干燥后得到粗提物,用于含量测定。
1.2.2纯化取预处理好的AB-8 大孔树脂20 mL,装入径高比为1 ∶10 的玻璃层析柱中,加压使其牢固。 将CR 提取后所得的浓缩液沿壁缓慢加入,上样量为5 mL,吸附1 h。 以40 mL/h 的流速,用120 mL 水充分洗脱除杂。 依次使用30%、50%和80%的乙醇各120 mL 进行洗脱,合并洗脱液(共计360 mL),以20 mL 为单位收集,进行黄酮和多酚含量测定以确定合适的洗脱液。 将洗脱液收集,减压浓缩后真空干燥,得到纯化样品,干燥器内储存备用。
1.3 活性成分含量测定
1.3.1CR 提取物中总黄酮 按照文献[15]方法,精密称取芦丁对照品5 mg,加入80%乙醇溶液,超声波溶解,定容,制得1 g/L 的芦丁对照品溶液。 精密称取5 mg 纯化样品作为供试品,加入80%乙醇溶液,超声波溶解,定容,制得1 g/L 的供试品溶液。 精密称取亚硝酸钠、硝酸铝和氢氧化钠,分别加入蒸馏水溶解,定容,制得5%亚硝酸钠溶液、10%硝酸铝溶液和4%氢氧化钠溶液。
分别准确移取10、50、100、150、200、250 和300 μL 的芦丁对照品溶液,以80%乙醇补充至0.5 mL。加5%亚硝酸钠溶液0.15 mL,静置6 min,加10%硝酸铝溶液0.15 mL,静置6 min,加4%氢氧化钠溶液2 mL,加蒸馏水2.2 mL,混匀,静置3 min。 测定508 nm 吸收波长下的吸光度。 以对照品质量浓度为横坐标,吸光度为纵坐标绘制标准曲线。 准确移取250 μL 供试品溶液,同上操作,每种供试品平行制备3份,进行总黄酮含量测定。 取其中一份供试品重复测量6 次考察仪器精密度,同时每隔10 min 测量一次考察样品稳定性。
1.3.2CR 提取物中总多酚 按照文献[16]方法,精密称取没食子酸对照品5 mg,加入蒸馏水,超声波溶解,定容,制得0.112 g/L 的没食子酸对照品溶液。 精密称取5 mg 纯化样品作为供试品,加入蒸馏水,超声波溶解,定容,制得1 g/L 的供试品溶液。 精密称取碳酸钠,加入蒸馏水溶解,定容,制得20%碳酸钠溶液。
分别准确移取50、100、150、200、250 和300 μL 的没食子酸对照品溶液,以蒸馏水补充至3 mL,加入福林酚试剂250 μL,静置5 min,加入20% Na2CO3溶液0.75 mL,混匀,静置30 min,测定763 nm 吸收波长下的吸光度。 以对照品质量浓度为横坐标,吸光度为纵坐标绘制标准曲线。 准确移取250 μL 供试品溶液,同上操作,每种供试品平行制备3 份。 精密度、稳定性考察同1.3.1节操作。
1.3.3HPLC 法同时测定CR 提取物的主要化学成分 分别精密称取儿茶素、原儿茶酸、香豆素、肉桂酸、肉桂醛和o-甲氧基肉桂醛6 种对照品,用50%甲醇配制成1 g/L 的对照品母液。 6 种对照品各取20 μL,混合均匀,补充280 μL 50%甲醇得到各对照品质量浓度为50 mg/L 的混合对照品母液,梯度稀释至20、10、4、2、1、0.4 和0.2 mg/L,过滤膜以供HPLC 测量分析,以对照品浓度为横坐标,吸光度为纵坐标绘制标准曲线。 精密称取纯化样品作为供试品,加入50%甲醇,制得1 g/L 的供试品溶液,过滤膜以供HPLC测量分析。 取其中一份供试品重复测量6 次考察仪器精密度,同时每隔8 h 测量一次考察样品48 h 内稳定性。
HPLC 条件:Inertsustain C18柱(4.6 mm ×150 mm,5μm);流动相为0.1%甲酸水溶液(A)和乙腈(B)。 洗脱梯度:0 ~2 min 10% B;2 ~10 min 10% ~20% B; 10 ~30 min 20% ~60% B; 30 ~32 min 60% ~10% B;32 ~37 min 10% B。
1.4 抗氧化能力测定
1.4.1总抗氧化能力按照文献[17]方法,采用总抗氧化能力(T-AOC)测定试剂盒测定供试品和对照品总抗氧化能力,使用蒸馏水分别配制不同质量浓度的纯化样品和抗坏血酸(Vc)溶液,质量浓度为10 ~2 000 mg/L,按照T-AOC 测定试剂盒说明书进行操作。 在520 nm 测定样品吸光度(A1)和空白对照品吸光度(A2),根据式(1)计算总抗氧化能力(η总)。
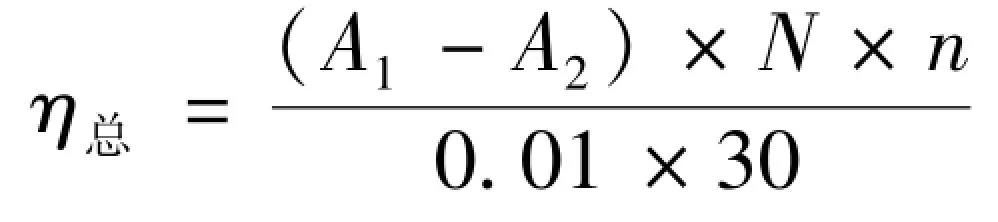
(1)
式中:N—反应体系稀释倍数(反应液总量/取样量);n—样品测试前稀释倍数。
在37 ℃时,每分钟每毫升样品使反应体系的吸光度值每增加0.01 时,为一个总抗氧化能力单位。
1.4.2DPPH·清除能力按照文献[18]方法,取不同质量浓度的供试品和对照品溶液(或蒸馏水)0.5 mL,质量浓度10 ~2 000 mg/L,加入0.5 mL DPPH 乙醇(或乙醇)溶液,混匀,室温放置30 min,以Vc 溶液作为阳性对照,测定517 nm 处的吸光度。 根据式(2)计算DPPH·清除率。
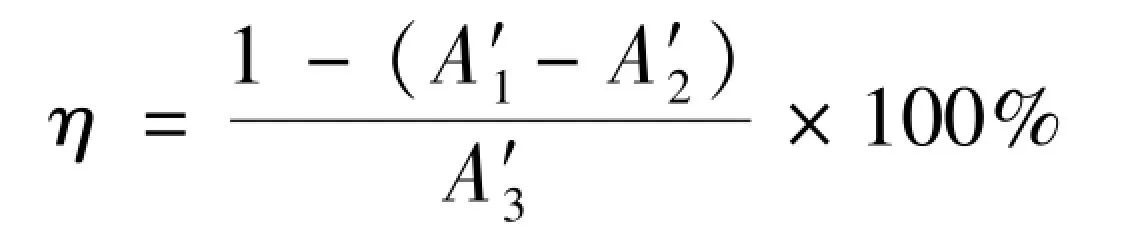
(2)
式中:η—自由基清除率,%;A′1—样品吸光度;A′2—样品本底吸光度;A′3—空白吸光度。
1.4.3·OH 清除能力 按照文献[19]方法,取2 mmol/L 水杨酸钠/乙醇溶液0.2 mL,加入9 mmol/L硫酸亚铁溶液(或蒸馏水)0.2 mL,分别加入0.6 mL 不同质量浓度的对照品和供试品(或蒸馏水)0.6 mL,最后加入6 mmol/L 双氧水0.2 mL,37 ℃反应1 h,以Vc 溶液作为阳性对照,在510 nm 下测定吸光度。 根据式(2)计算·OH 清除率。
1.5 数据处理
实验数据以mean±SD 表示,平行实验≥3 次,并使用SPSS19.0 软件进行方差分析和检验。
2 结果与讨论
2.1 黄酮与多酚含量测定的方法学考察
2.1.1黄酮含量测定以NaNO2-Al(NO3)3-NaOH 为显色剂,采用UV 分光光度计测定样品中总黄酮的含量。 方法学考察包括线性、精密度和稳定性。 以芦丁为对照品绘制了标准曲线,结果表明:在质量浓度1.28 ~38.34 mg/L 范围内,溶液质量浓度与508 nm 处的吸光度的线性关系良好,回归方程为y=9.941 7x-0.007 5,R2=0.991 3。 使用任意一份样品重复测量6 次,吸光度在0.218 69 ~0.218 96 之间,相对平均偏差(RSD)为0.05%,表明仪器精密度良好。 将同一份样品每隔10 min 测量一次,考察了样品50 min 以内的稳定性,6 次测定的吸光度在0.208 16 ~0.218 72 之间,RSD 值为1.73%,表明显色反应后溶液能在50 min 内保持稳定。 由此可知,总黄酮含量测定方法的线性、精密度和稳定性良好。
2.1.2多酚含量测定以福林酚比色法测定样品中总多酚的含量,并对多酚含量的测定进行方法学考察。 以没食子酸为对照品绘制了标准曲线,结果表明:在质量浓度1.40 ~8.40 mg/L 范围内,溶液质量浓度与763 nm 处的吸光度线性关系良好,回归方程为y=86.822x+0.012 8,R2=0.999 9。 6 次重复测量的吸光度在0.510 14 ~0.510 61 之间,RSD 值为0.04%。 50 min 溶液稳定性测定的吸光度在0.509 84 ~0.515 58 之间,RSD 值为0.44%,表明显色反应后溶液能在50 min 内保持稳定。 由此可见,总多酚含量测定方法的线性、精密度和稳定性良好。
2.2 CR 提取物活性成分的提取、纯化及含量测定
2.2.1活性成分的提取与纯化肉桂在水蒸气蒸馏法提取过程中许多非挥发性的成分会溶解到水中,因此保留了水蒸气蒸馏提取过程的水提液。 虽然多酚类和黄酮类成分中的黄酮苷极性较大,易溶于水,但是槲皮素、山奈酚等黄酮类以及部分多酚类成分在有机溶剂中的溶解度远远超过水溶液。 为了对CR的化学成分提取更加全面,本研究将CR 与水提液分离之后,以乙醇为提取溶剂,采用超声波辅助提取法对CR 进行二次提取。
将提取液合并、浓缩、真空干燥之后所得样品中多酚和黄酮含量普遍偏低,两次提取之后,LR、TR 和BR 粗提物中黄酮质量分数分别为4.31%、15.05%和8.53%;LR 粗提物中多酚质量分数低于定量限,在TR 和BR 粗提取物中多酚质量分数分别为8.24%和7.16%。 由此可见,CR 的不同部位经水提和醇提之后,提取物中存在黄酮和多酚类成分,但是含量较低。 已有文献报道肉桂的主要活性成分为黄酮和多酚,为了提高粗提物中活性成分的含量,选择AB-8 大孔树脂对粗提物中的黄酮和多酚进行纯化。 为了对活性成分进行特异性洗脱,依次用30%、 50%和80%的乙醇溶液各120 mL 进行洗脱,将不同阶段收集到的洗脱液真空干燥之后分别测定多酚和黄酮含量,绘制洗脱曲线,结果见图1。 由图1 可知,30%乙醇溶液对黄酮和多酚类成分几乎没有洗脱能力,当乙醇体积分数提高到80%时,对黄酮和多酚的洗脱能力最强,之后的洗脱液中几乎没有黄酮和多酚类成分,因此选用80%的乙醇作为洗脱溶剂。
2.2.2纯化前后黄酮和多酚含量变化采用大孔树脂对提取液充分吸附之后,使用80%的乙醇溶液进行洗脱,洗脱液真空浓缩干燥得到的样品中黄酮和多酚的含量显著提高(见表1)。 以LR 为原料,纯化之后提取物中黄酮质量分数从4.31%提高到18.50%,多酚质量分数从低于定量限提高到2.60%。 以TR 为原料,黄酮质量分数从15.05%提高到53.94%,多酚质量分数从8.24%提高到37.56%。 以BR 为原料,黄酮质量分数从8.53%提高到44.48%,多酚质量分数从7.16%提高到28.16%。 3 种原料的提取物中黄酮均高于多酚;两者总含量在3 种原料的提取物差异较大,在TR 和BR 提取物中的质量分数超过50%,分别为91.49%、72.64%,在LR 提取物中的质量分数为21.10%。
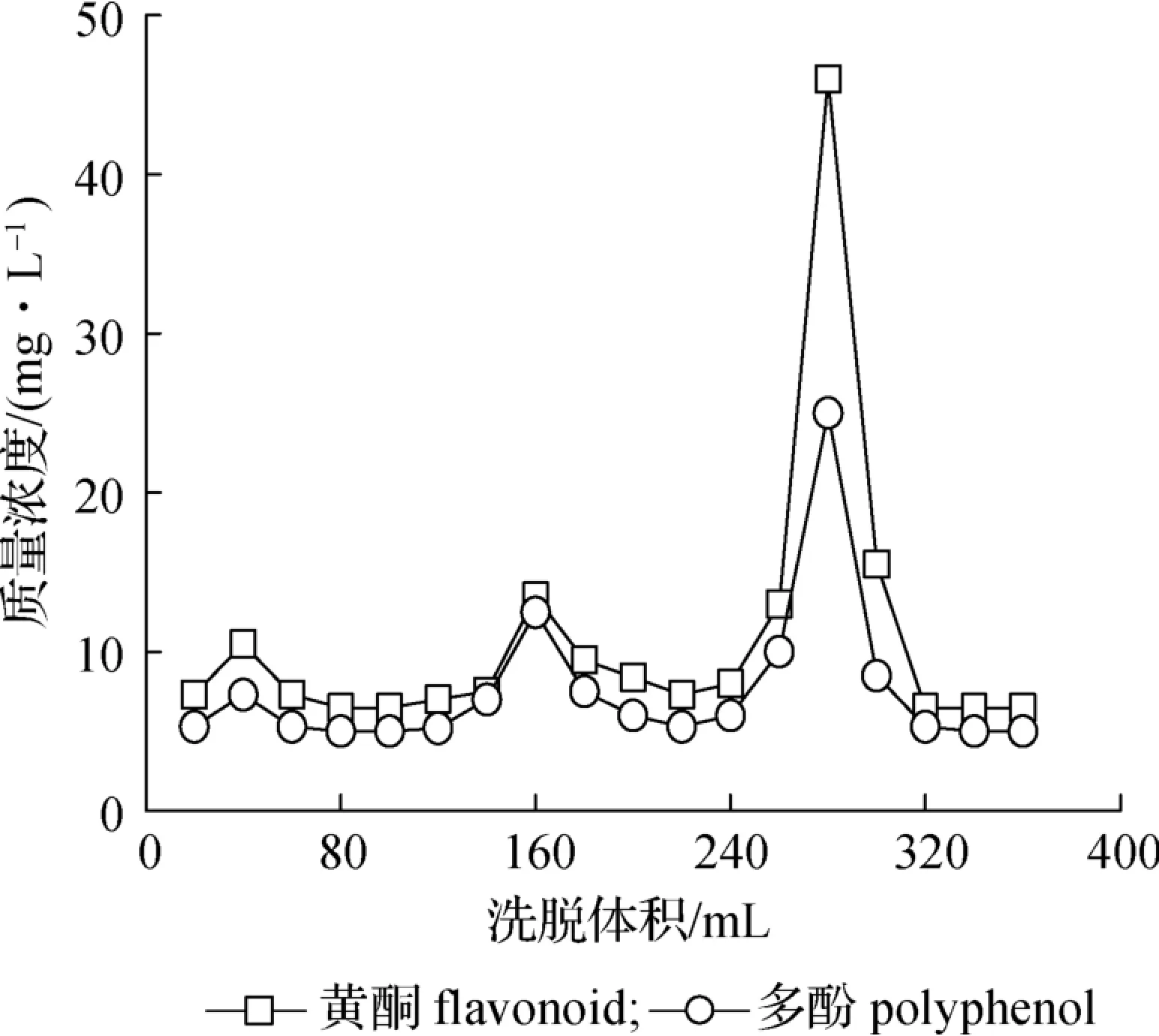
图1 AB-8 树脂对黄酮和多酚的洗脱曲线Fig. 1 The elution curves of flavonoid and polyphenol by AB-8 resin
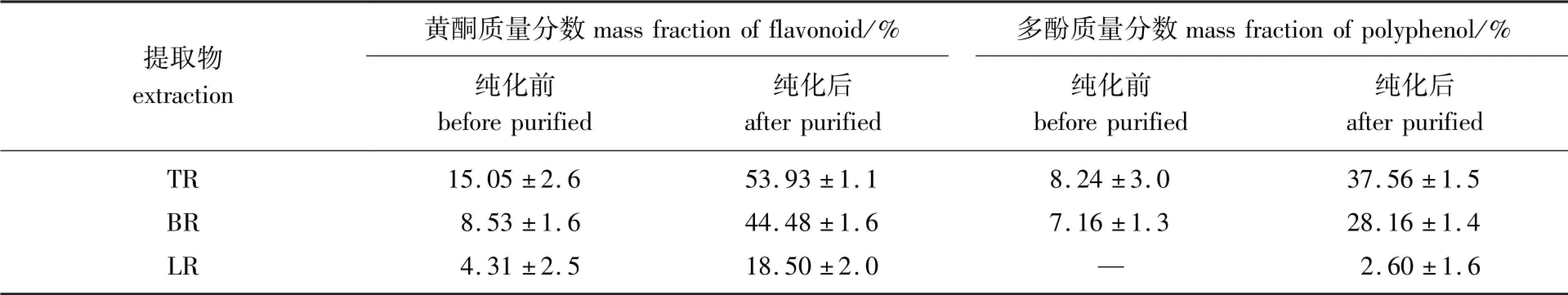
表1 肉桂蒸油剩余物的提取物中黄酮和多酚的含量Table 1 The contents of flavonoid and polyphenol extracted from cinnamon distillation residue
2.3 HPLC 测定主要成分
借助HPLC 对CR 提取物的多酚、黄酮和其他主要成分进行了精确的定量分析。 研究对象包括原儿茶酸、儿茶素、香豆素、肉桂酸、肉桂醛和o-甲氧基肉桂醛,这些化合物结构中均含有多个B 环取代羟基,因此具有一定的还原性[3]。 首先比较了6 种对照品在220 ~380 nm 波长下的色谱图,为了能够对主要成分在同一波长下检测,最终选择在270 nm 波长处进行紫外检测,HPLC 色谱图见图2。 通过比较6 种对照品的出峰时间和提取物中主要色谱峰的出峰时间,确定了CR 提取物中多酚类成分有原儿茶酸和儿茶素,对应的出峰时间分别为7.07 和10.23 min,黄酮和其他成分有香豆素、肉桂酸、肉桂醛和o-甲氧基肉桂醛,出峰时间分别为21.03、23.41、25.71 和27.85 min。 这6 种成分为CR 提取物中含量较高的成分,可能是蒸油不充分所致,因此在色谱图中出现了较强的色谱峰,其他多酚和黄酮类成分如没食子酸、芦丁等可能由于含量较低,没有在色谱图中明显出峰。
样品的HPLC 色谱图中6 种成分的分离度>2.0,因此可以实现准确定量。 以峰面积为纵坐标,对照品质量浓度为横坐标绘制出6 种对照品的标准曲线,结果见表2。
6 条校正曲线的R2均>0.999,在相应的定量范围内可用于样品中主要成分的含量计算。 6 种成分重复测量结果的RSD 值在0.43% ~14.07%之间,均<15%,表明仪器精密度良好;48 h 内6 种成分稳定性测定结果的RSD 值在0.19% ~13.90%之间,均<15%,表明待测溶液能在48 h 内保持稳定。 因此,HPLC 测定方法的线性、精密度和稳定性良好,适用于6 种成分的含量测定。
6 个主成分的含量测定结果见表2。 由表2 可以看出:3 种原料对应的提取物中含量最高的成分为香豆素或肉桂酸,属于黄酮类成分。 6 个主成分在TR、LR 和BR 的提取物中含量差异较大,含量由高到低的顺序为TR >BR >LR,这与显色法测定提取物中总黄酮和总多酚的结果一致。
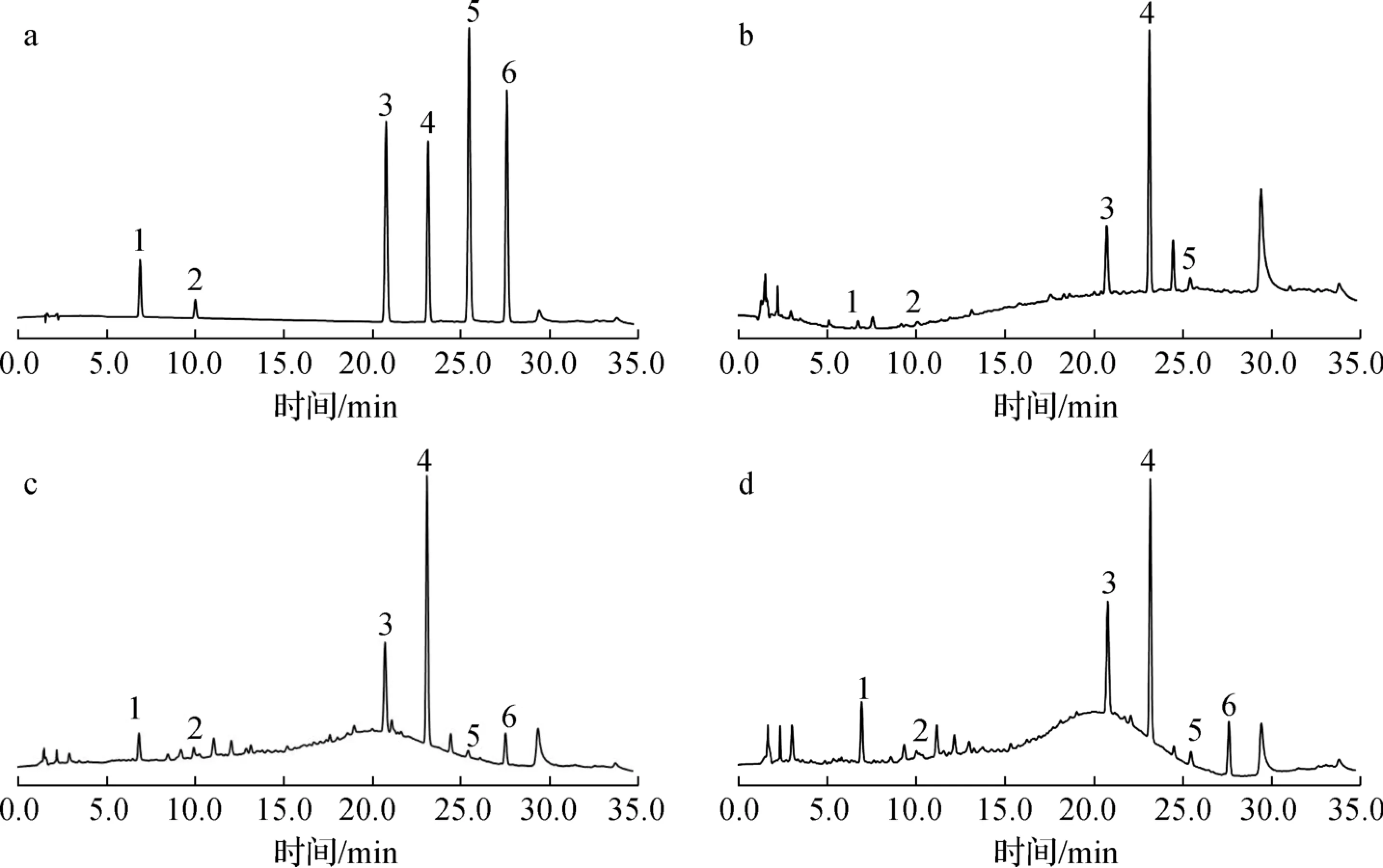
1.原儿茶酸protocatechuic acid;2.儿茶素catechin;3.香豆素coumarin;4.肉桂酸cinnamic acid;5.肉桂醛cinnamic aldehyde;6.o-甲氧基肉桂醛methoxycinnamaldehydea.对照品contral; b.LR; c.TR; d.BR
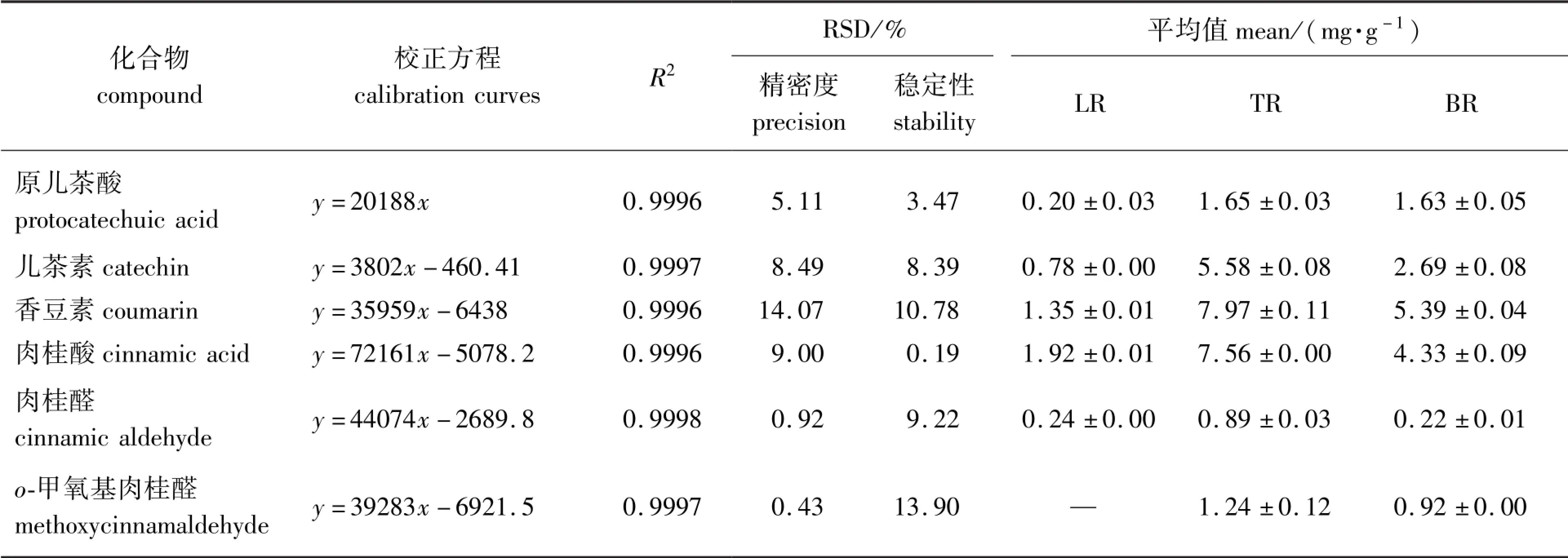
表2 HPLC 测定主成分含量结果Table 2 The contents of main compounds measured by HPLC
2.4 抗氧化活性
2.4.1总抗氧化能力3 种原料的提取物及Vc 在不同质量浓度时的总抗氧化能力见图3(a)。 由图3(a)可见,所有提取物的总抗氧化能力都随着溶液质量浓度的增加而增强,与Vc 趋势相同。 TR 和BR提取物的总抗氧化能力在质量浓度为0.2 g/L 时达到最大,分别为8.2 和8.0 U/mL。 TR 和BR 提取物的总抗氧化能力达到了Vc 的60%以上;LR 的总抗氧化能力相对较差,约为Vc 总抗氧化能力的40%。
2.4.2DPPH·清除能力如图3(b)所示,随着提取物和Vc 质量浓度的增加,各种物质对DPPH·的清除率均表现为先增加后趋于稳定。 质量浓度低于0.1 g/L 时,Vc、TR 和BR 提取物的DPPH·清除能力随质量浓度的增加而逐渐增加;而LR 提取物的DPPH·清除能力在质量浓度低于0.2 g/L 的范围内随质量浓度的增加而逐渐增加。 计算各物质的半数抑制浓度(IC50)值,结果表明:Vc、TR、BR 和LR 提取物的IC50值分别为0.012、0.017、0.021 和0.150 g/L。 由此可知,TR 和BR 提取物对DPPH·的清除效果较好,清除率略低于Vc,与总抗氧化能力的结果一致。
2.4.3·OH 清除能力 如图3(c)所示,当质量浓度低于0.05 g/L 时,Vc 的·OH 清除率随质量浓度的增加而逐渐增加;当质量浓度低于0.2 g/L 时,TR 提取物的·OH 清除率随质量浓度的增加而逐渐增加,其IC50值为0.12 g/L。 当·OH 清除率趋于稳定时,TR 提取物对·OH 的清除率达到Vc 的80%左右,BR和LR 提取物对·OH 的清除能力相对较差,对·OH 的清除率约为Vc 的40% ~50%。
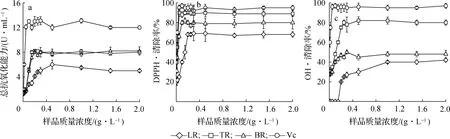
a.总抗氧化能力total antioxidant capability; b.DPPH·; c.·OH图3 肉桂蒸油剩余物提取物的体外抗氧化性能Fig. 3 Thein vitroantioxidant properties of extracts from cinnamon distillation residue
3 结 论
3.1利用显色反应分别建立了肉桂蒸油剩余物(CR)提取物中总黄酮和总多酚的含量测定方法,不仅操作简便快速,而且相对于盐酸镁粉法更加安全可靠。 系统的方法学考察结果表明:线性相关系数R2均>0.99,精密度RSD <0.05%,稳定性RSD <2.00%,适用于总黄酮和总多酚的含量测定。 6 种主成分的HPLC 分析方法线性相关系数R2均>0.999,精密度和稳定性RSD <15.00%,适用于样品含量测定。
3.2利用超声波辅助乙醇提取法对CR 进行二次提取,并与蒸汽蒸馏提取后的水溶液合并,使用AB-8大孔树脂进行纯化,使样品中黄酮和多酚的含量提高了2.6 ~4.2 倍。 LR、TR、BR 提取物中总黄酮含量均大于总多酚;黄酮和多酚在TR 和BR 提取物中的总质量分数分别为91.49%、72.64%,在LR 提取物中的总质量分数为21.10%;3 种原料对应的提取物中含量最高的成分为香豆素或肉桂酸; 6 个主成分在TR、LR 和BR 提取物中含量差异较大,含量由高到低的顺序为TR >BR >LR。
3.33 种原料对应提取物的抗氧化能力与样品质量浓度有较强的相关性。 TR 和BR 提取物的总抗氧化能力在质量浓度为0.2 g/L 时达到最大,分别为8.2 和8.0 U/mL;TR、BR 和LR 提取物清除DPPH·的IC50值分别为0.017、0.021 和0.150 g/L,略高于Vc 的0.012 g/L;TR 提取物对·OH 的清除率达到Vc 的80%,而BR 和LR 提取物对·OH 的清除率相对较差,约为Vc 的40% ~50%。
猜你喜欢
中老年保健(2022年2期)2022-08-24
中老年保健(2021年5期)2021-12-02
四川蚕业(2021年3期)2021-02-12
中国调味品(2020年7期)2020-07-24
中国调味品(2019年2期)2019-03-18
现代营销(创富信息版)(2018年2期)2018-08-15
中成药(2017年8期)2017-11-22
中成药(2017年10期)2017-11-16
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
