
抗结核药物的研究进展
2020-05-11 07:43韦媛媛于丽芳
中国药科大学学报 2020年2期
韦媛媛,杨 帆,汤 杰,于丽芳
(华东师范大学化学与分子工程学院,上海分子治疗与新药创制工程技术研究中心,上海 200062)
结核病(tuberculosis,TB)是一种在全球范围内流行的慢性致死传染性疾病,由结核分枝杆菌(mycobacterium tuberculosis,Mtb)引起。结核病主要通过呼吸道传播,通常会影响肺部,也可能会影响身体的其他部位,如大脑、肾和脊柱等。如果得到良好的治疗,大多数情况下结核病可以被治愈,但是如果治疗不恰当则可能产生严重后果,甚至导致死亡。2018年世界卫生组织评估报告显示,结核病是世界范围内导致死亡的十大原因之一,也是单一传染因子致死率最高的疾病。2017年,全球有近1 000万结核病患者,其中160万人死于结核病。更令人担忧的是,Mtb的耐药问题仍然没有得到有效解决。多药耐药结核病(MDR-TB)是由至少对异烟肼和利福平都耐药的Mtb引起的,而这两者是目前最有效的抗结核药物,基本用于治疗所有的结核病患者。而引起广泛耐药结核病(XDR-TB)的Mtb除了对异烟肼和利福平耐药,还对氟喹诺酮类药物或可注射的3种二线药物之一耐药,因此在大多数情况下XDR-TB无法治愈。据报道,2017年约558 000人患有对利福平耐药的结核病,其中有82%是MDR-TB,而8.5%的MDR-TB是XDR-TB。在全球范围内,3.5%的新增患者以及18%的先前治疗过的患者患有对利福平耐药的结核病[1]。而HIV感染者因为免疫能力弱,更容易患病,死亡风险更高。
目前,对药敏性结核病的推荐疗法是4种一线药物(异烟肼、利福平、吡嗪酰胺和乙胺丁醇)联用6个月的方案,治疗成功率高于85%,而全球MDR-TB的治疗成功率只有55%,因此MDR-TB的治疗已成为全球关注的焦点。对利福平耐药的结核病以及MDR-TB的治疗需要使用不良反应更大的药物,花费更多费用(≥1 000美元/人)[2],而且MDR-TB的治疗时间长达20个月以上,这给患者对治疗方案的依从性带来极大挑战[3]。随着现有药物耐药率的不断提高,针对MDR-TB的新药需求也越来越迫切。本文将目前临床上的12个新化学实体按照作用机制(靶点如图1所示)分成3类:靶向细胞壁合成、靶向能量代谢以及靶向蛋白质合成,并分别进行综述,旨在为今后的抗结核药物研究提供参考。
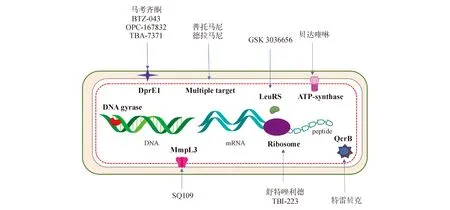
图1 抗结核新药的靶点
1 靶向细胞壁作用合成
抑制Mtb细胞壁的生物合成是一个有效的抗结核药物开发策略,一线药物异烟肼和乙胺丁醇均是靶向细胞壁合成的过程。Mtb的细胞壁由独特的内层和围绕质膜的外层组成,内层由肽聚糖(PG)、阿拉伯半乳聚糖(AG)和分枝菌酸(MA)构成,它们共价连接形成MA-AG-PG复合物。该复合物能够形成疏水的渗透性屏障,阻止许多环境溶剂进入,从而使Mtb耐酸,并能够在极端恶劣的环境下生存,因此被认为是Mtb细胞壁的核心结构[4],是抗Mtb药物的重要靶标。
Mtb在宿主中生存依赖于其细胞壁的低渗透性,因此,参与细胞壁不同组分生物合成以及这些组分附着所必需的酶都是潜在的药物靶标[5]。
1.1 德拉马尼(delamanid,OPC-67683)
德拉马尼是大冢制药研发的一种硝基咪唑类杀菌药,它的作用机制为抑制分枝菌酸的合成。其于2014年获得欧盟委员会的上市批准,在患者对基本治疗药物耐药以及对其他治疗方案不耐受的情况下,该药可作为联合治疗方案的一部分,用于成人MDR-TB的治疗[6]。
德拉马尼对H37Rv菌株的MIC为0.012 μg/mL[7],在体内低剂量就有较高的抗结核活性。口服德拉马尼4~8 h后达到药峰浓度,10~14 d达到稳态,血浆半衰期为30~38 h,大部分德拉马尼及其代谢产物通过粪便排泄。H37Rv菌株对德拉马尼的自发抗性频率为6.44×10-6~1.22×10-5,与异烟肼相当[8]。德拉马尼对CYP450酶无抑制或诱导作用,所以它能够与其他药物联用,包括一些会诱导CYP450酶或者由CYP450酶代谢的抗HIV药物。
临床试验显示,接受每日两次的100 mg德拉马尼联合优化的背景方案治疗2个月的患者,痰培养转阴的比例显著高于接受安慰剂组的比例(45.4%vs29.6%)。值得一提的是,在接受德拉马尼治疗的组中,虽QT间期延长发生的频率更高,但是没有出现因QT间期延长而发生的临床事件(如晕厥、心律不齐等)[9]。此外,临床研究以外的最新结果显示,在常规的治疗方案中加入德拉马尼对高度耐药性结核病患者(包括XDR-TB)的效果较好[10]。
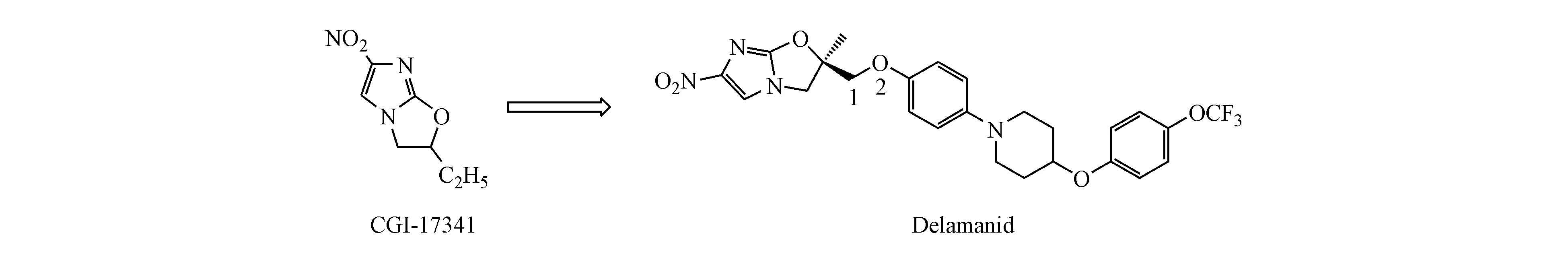
图2 德拉马尼(delamanid)的发现
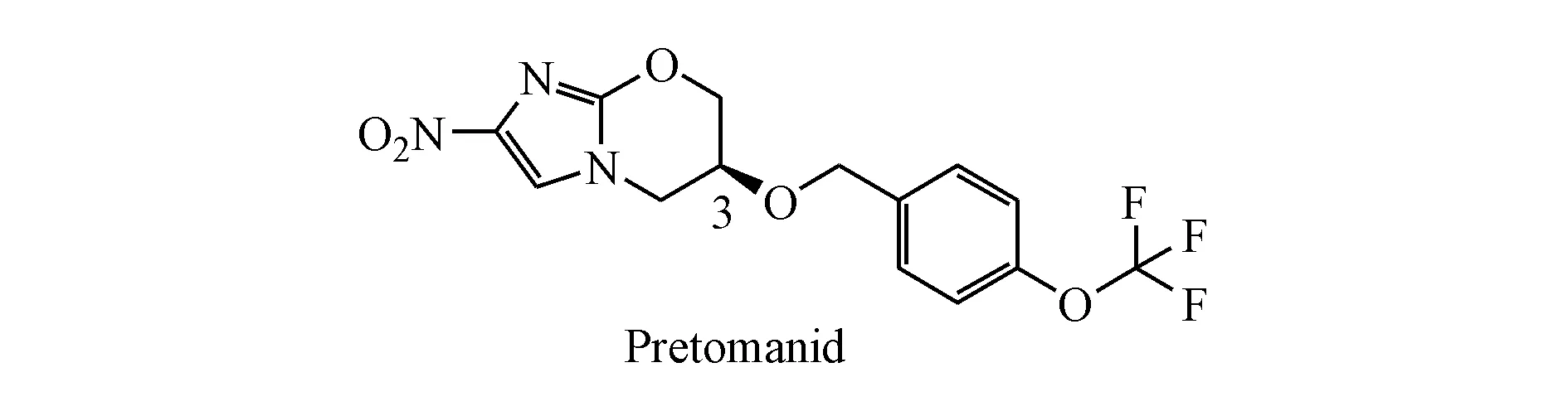
1.2 普托马尼(pretomanid,PA-824)
普托马尼也是一种硝基咪唑类化合物,最初由Pathogenesis公司发现临床有效,其作用机制是抑制Mtb蛋白质和霉菌酸的合成。2019年8月14日,FDA批准普托马尼与贝达喹啉和利奈唑胺联合使用来治疗XDR-TB以及对其他疗法不耐受或无响应的MDR-TB。与目前已有的抗结核药物不同,普托马尼对处于复制期和非复制期的Mtb均有杀菌活性[11],其代谢过程中能够产生活性氮物质(如一氧化氮),是Mtb的呼吸毒素,有助于对非复制期Mtb的杀灭[12]。
Stover等[11]借鉴CGI-17341结构合成了328个3位取代的硝基咪唑并吡喃类化合物,研究发现C3位引入亲脂性取代基对活性有利,此外该位置的立体化学对活性影响很大,S构型比R构型活性至少高10倍。其中普托马尼对敏感菌株以及多药耐药菌株的MIC为0.031~0.531 μg/mL[13]。它对多药耐药菌株与敏感菌株表现出相似的敏感性,表明其与现有的抗结核药物没有交叉耐药性。在小鼠模型中,普托马尼在治疗的初始和持续阶段均具有剂量依赖性,在100 mg/(kg·d)的剂量下,其杀菌活性接近异烟肼,对于人类的等效剂量为25 mg/(kg·d)[14]。在14 d抗结核研究中,普托马尼与莫西沙星和吡嗪酰胺组合的活性不低于目前临床上使用的异烟肼、利福平、乙胺丁醇和吡嗪酰胺的标准药物组合。因为这种药物组合不依赖异烟肼和利福平,所以也可以用来治疗对二者有耐药性的结核病患者[15]。多项研究表明,普托马尼与很多其他抗结核药物的联用有很好的效果[16-18]。
普托马尼在基因毒性研究中没有显示出致突变作用,也没有明显的CYP450相互作用,并且对多数革兰阳性和阴性细菌没有显著活性,表现了令人满意的微生物选择性。
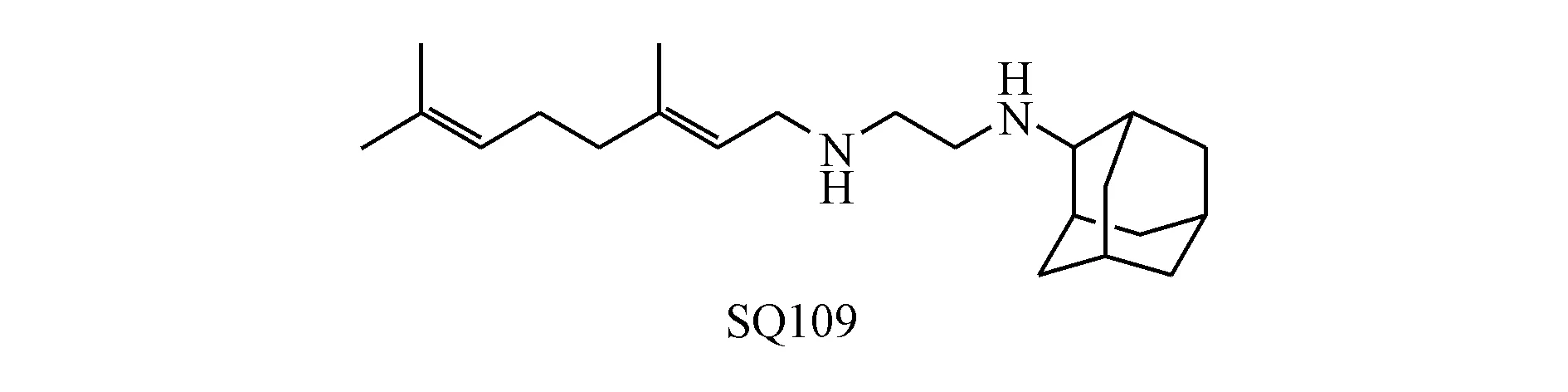
1.3 SQ109
SQ109是一种新型的乙二胺类小分子药物,目前处于临床Ⅱ期,它靶向Mtb的膜蛋白MmpL3(Mycobacterial membrane protein Large 3),而MmpL3是将分枝菌酸转运到细胞壁所需的转运蛋白。
1999年,Sequella和美国国立卫生研究院过敏和传染病研究所对63 238个化合物进行了合成和筛选(这些化合物都含有在乙胺丁醇中发现的1,2-乙二胺药效团),得到了170个MIC小于6 μg/mL的化合物。同时考虑化合物的细胞毒性、Mtb感染的巨噬细胞中的杀菌活性以及化合物的亲脂性,选出11个化合物进行小鼠体内的药效研究,对其中最有开发前景的3个化合物进行了小鼠最大耐受剂量以及药代动力学的研究。其中表现最好的SQ109不仅对敏感菌株H37Rv有活性(MIC≤0.2 μg/mL),而且对于临床上分离的MDR-TB以及XDR-TB菌株也有活性。它可以杀死巨噬细胞内的Mtb,其作用优于乙胺丁醇,和异烟肼相当,在最小抑菌浓度下,可以使细胞内Mtb降低99%[19]。其与利福平、异烟肼以及舒特唑利德均有协同作用[20-22]。
2012- 2016年间,俄罗斯联邦6个研究中心评估了在MDR-TB的标准治疗方案中使用SQ109的有效性和安全性。结果显示:在化疗强化阶段的第6个月末,与安慰剂组相比,SQ109治疗组有更多患者的细菌停止排泄,两组间的痰转化结果无统计学差异,但在第8周结束时,SQ109治疗组的痰转化为52%,安慰剂组为38%;SQ109治疗组细菌停止排泄的中位时间为56 d,安慰剂组为84 d;与安慰剂加基本抗结核治疗组相比,SQ109与治疗MDR-TB的基础化疗药物合用不会导致更多的不良事件。所以,SQ109作为肺部MDR-TB化疗方案的一部分,有效且耐受良好。
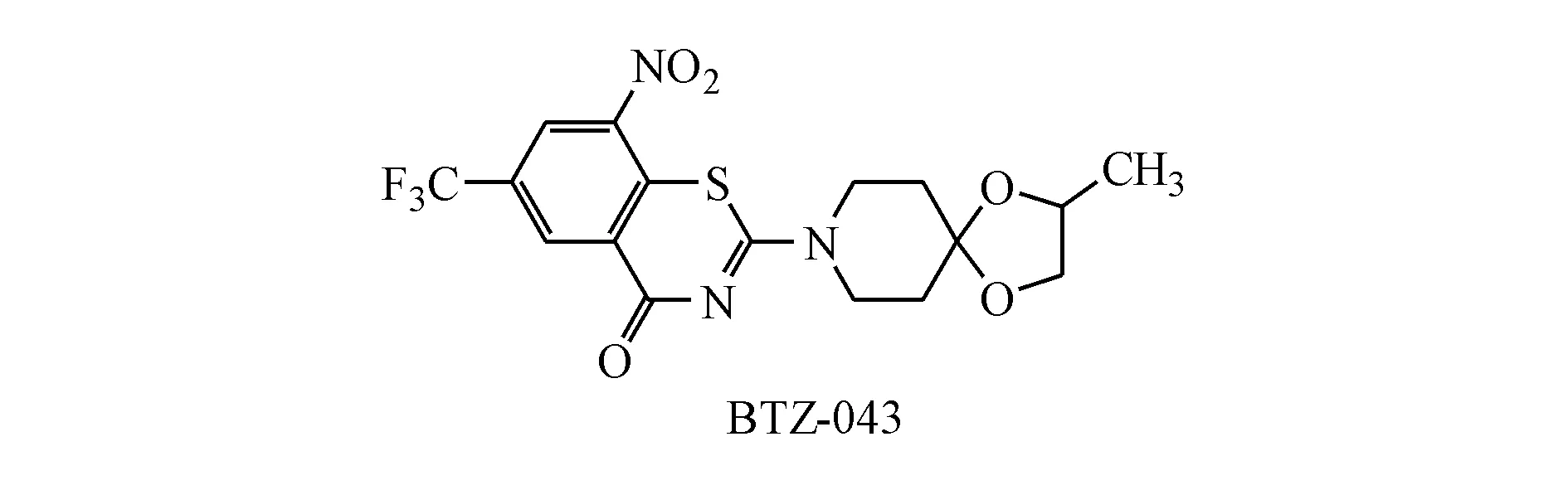
1.4 BTZ-043
BTZ-043是苯并噻嗪酮的衍生物,目前处于临床Ⅰ期,其作用机制是通过共价抑制DprE1(decaprenyl-phospho-β-D-ribose oxidase)从而抑制Mtb细胞壁的合成。DprE1与DprE2组成异二聚体酶DprE,它参与十聚异戊烯磷酰基-β-D-阿拉伯呋喃糖(DPA)的生物合成,而DPA是细胞壁生物合成所必须的阿拉伯糖的唯一供体[23]。DprE1因其必需性和细胞质外的定位被认为是Mtb致命弱点[24]。
Makarov等[25]对一系列含硫杂环化合物的活性筛选中发现硝基苯并噻嗪酮类化合物对分枝杆菌有很好的活性,其中BTZ-043对Mtb H37Rv菌株的MIC为1 ng/mL,对测试的其他菌株也有高效的抑制活性,包括从临床分离的MDR和XDR菌株。它72 h内可以将细菌体外活力降为原来的千分之一,与异烟肼杀菌效果相当。BTZ-043在小鼠体内试验中效果优于异烟肼,治疗时间超过2个月效果尤为显著。
BTZ-043与异烟肼、乙胺丁醇、普托马尼、莫西沙星、美罗培南以及SQ-109都没有拮抗作用,与利福平和贝达喹啉有协同作用[26]。临床前毒理学研究中发现BTZ-043毒性较低,28 d大鼠耐受水平高达170 mg/kg,小型猪耐受水平达到360 mg/kg。此外,其与CYP450酶相互作用较低,提示其可能能够与抗HIV药物联用。
1.5 马考齐酮(macozinone,MCZ,PBTZ-169)
马考齐酮是哌嗪基苯并噻嗪酮衍生物,目前处于临床Ⅰ/Ⅱ期研究,它的靶点是DprE1。DprE1-马考齐酮复合物晶体结构表明马考齐酮在活性位点与Cys387形成加合物(图3),导致DprE1不可逆失活[27]。
BTZ-043在小鼠模型中的效果低于预期,故而通过在骨架上引入哌嗪基对其进行优化得到一类活性提高的苯并噻嗪酮类化合物。其中马考齐酮对Mtb H37Rv菌株的MIC为0.3 ng/mL[28],并且与贝达喹啉、氯法齐明、德拉马尼以及舒特唑利德均有协同作用[29-30]。

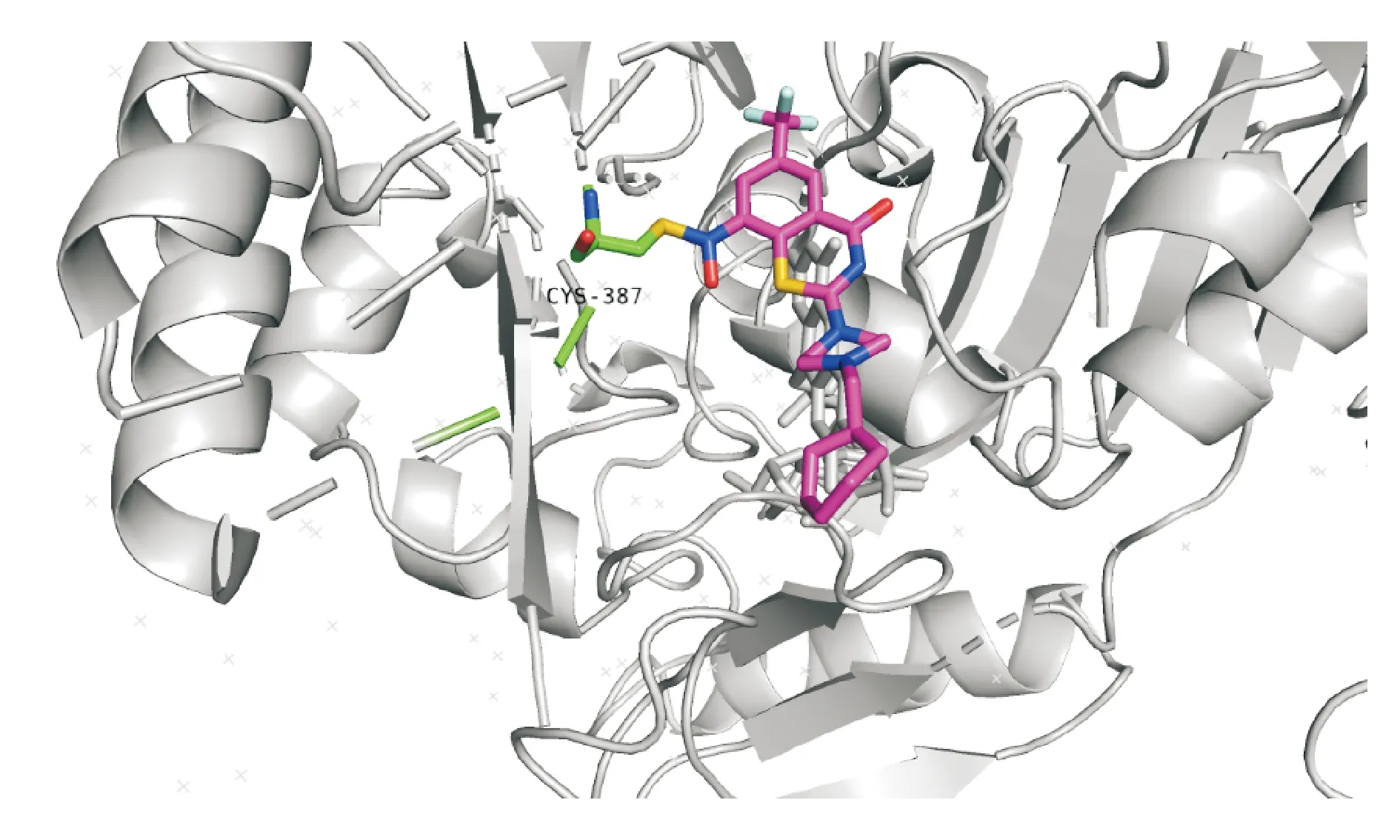
图3 马考齐酮在活性位点与Cys387形成加合物[27]
Cys387 (green sticks),Macozinone (purple sticks),PDB ID:4NCR
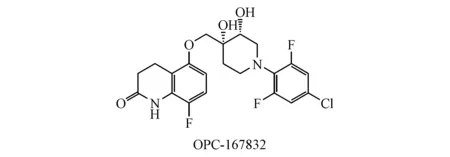
1.6 OPC-167832
OPC-16783是靶向DprE1的一种3,4-二氢卡司丁酮衍生物,目前处于临床Ⅰ期研究,其对实验室菌株以及临床分离耐药菌株的MIC范围为0.000 24~0.002 μg/mL。在慢性结核病实验小鼠模型中,其对生长期和细胞内Mtb均有杀灭活性。OPC-167832与德拉马尼等其他抗结核新药的联用比标准治疗方案更有效,具有缩短治疗时间,提高治疗效果的潜力[31]。
1.7 TBA-7371(AZ 7371)
TBA-7371属于氮杂吲哚类化合物,目前处于临床Ⅰ期,是DprE1的非共价抑制剂。TBA-7371的发现始于咪唑并吡啶类化合物1,它对H37Rv菌株的MIC为0.017 μmol/L,但最低杀菌浓度大于200 μmol/L。早期改造用吡咯并吡啶骨架代替咪唑并吡啶骨架得到化合物2,它的MIC为6.25 μmol/L,但最低杀菌浓度达到了12.5 μmol/L。采用骨架跃迁的策略用嘧啶环代替苯环得到了化合物3,但它存在代谢不稳定的问题,对其进一步优化得到代谢稳定的TBA-7371[32](图4)。其IC50为10 nmol/L,MIC为0.78~3.12 μmol/L。
在浓度高达100 μmol/L时对THP1(人单核细胞系)细胞没有抑制作用,在浓度高达33 μmol/L时对hERG通道亦没有抑制,表明其心血管毒性风险低。并且它对CYP450酶无抑制作用,有利于联合用药[32]。此外,它具有低相对分子质量、低lgD、优异的渗透性以及良好的口服暴露等优点。
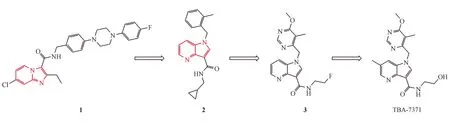
图4TBA-7371的发现
2 靶向能量代谢
特雷贝克和贝达喹啉均靶向Mtb的能量代谢过程,分别抑制细胞色素bc1和ATP合成酶,因而均能够降低Mtb的ATP产量,但当细胞色素bc1复合体被抑制时,需氧生长期细胞色素bd氧化酶就会参与能量的供给。特雷贝克正是由于存在替代的细胞色素bd氧化酶,使其表现出了较贝达喹啉更弱的杀菌活性[33-34]。
2.1 贝达喹啉(bedaquiline,TMC207)
贝达喹啉是美国强生公司研发的二芳基喹啉类药物,是40余年来第1个抗结核新药,它是分枝菌酸ATP合成酶抑制剂,而ATP合酶参与了Mtb能量供应过程。2012年,FDA批准贝达喹啉用于MDR-TB的治疗。2013年,当对其他治疗方案耐药或不耐受时,WHO推荐贝达喹啉用于MDR-TB的治疗。
强生公司针对耻垢分枝杆菌的全细胞筛选得到了化合物4(图5),接下来对化合物4的构效关系探索包括:改变季碳中心连接的侧链长度;用不同的胺以及非碱性基团代替侧链末端的二甲氨基;在侧链的第1个苯环上引入吸电子和给电子基团;用烷基、芳基以及杂环取代侧链的第2个苯环;在喹啉环不同位置引入不同取代基;以及测试了不同的异构体,最终发现了贝达喹啉[35]。
在临床Ⅱb试验中,相比于安慰剂组,贝达喹啉的MDR-TB治疗方案导致培养转化时间加快,培养转化率更高,治愈率更高。2010-2015年间,在对25个国家的12 000名MDR-TB患者的研究中发现,贝达喹啉有更高的治愈率和更低的病死率[36]。
与德拉马尼相似,贝达喹啉也有QT间期延长的风险。虽然鲜有因为使用贝达喹啉导致猝死的情况,但是患者应密切关注心电图测试以确保安全。
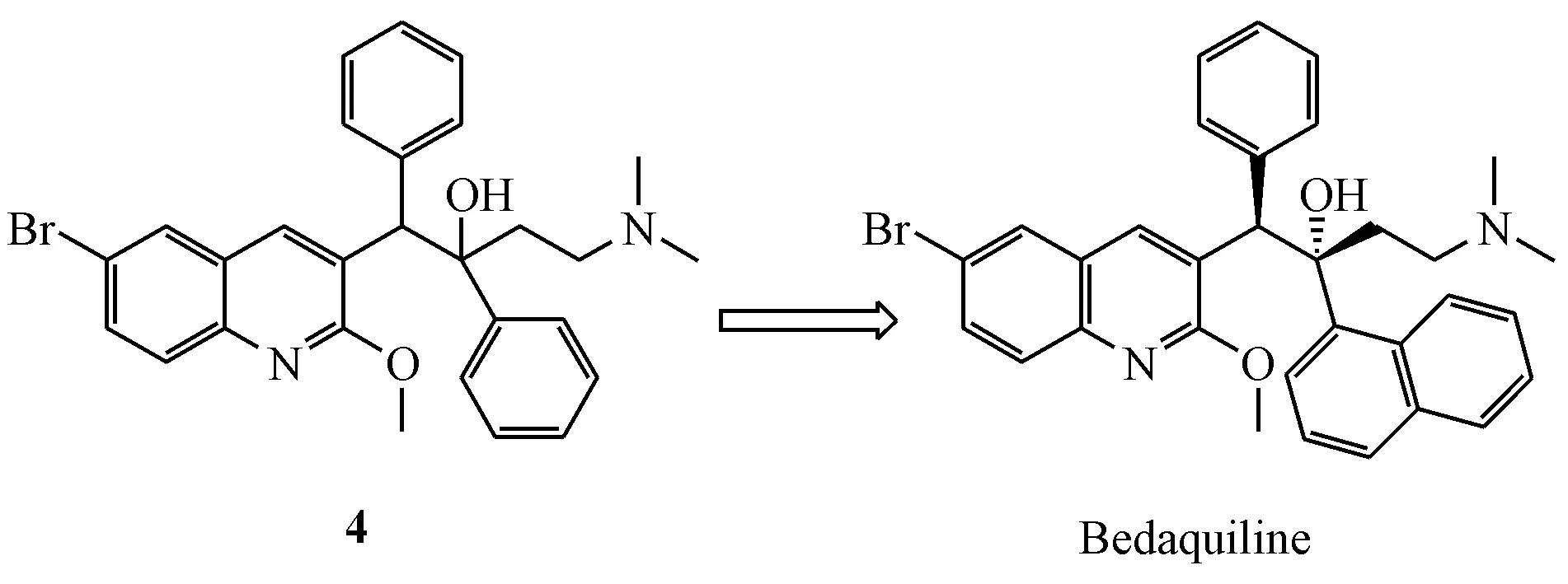
图5贝达喹啉(bedaquiline)的发现
2.2 特雷贝克(telacebec,Q203)
特雷贝克属于咪唑并吡啶酰胺类化合物,目前处于临床Ⅱ期研究,它通过与QcrB亚基结合抑制细胞色素bc1(电子传递链复合物Ⅲ)[37]。
特雷贝克的发现始于对多个商业化合物库的表型高内涵筛选,从121 156个化合物中得到了106个有活性的苗头化合物,其中的IPA01对巨噬细胞内、外的Mtb均有较好的活性(分别在1.25和1.86 μmol/L浓度下,抑制率达到50%),对IPA01的吡啶环、咪唑的2位甲基以及咪唑的3位侧链进行结构衍生合成了477个化合物并进行了评估,最终得到特雷贝克(图6)[38]。它能够干扰缺氧非复制性Mtb的ATP稳态,IC50小于10 nmol/L,在低浓度下可以快速抑制ATP合成。它在0.002 7 μmol/L浓度下对Mtb H37Rv菌株的抑制率为50%。在Mtb的急性小鼠模型中,给药剂量为10 mg/kg时,细菌负荷减少90%以上,而在慢性小鼠模型中,给药剂量分别为0.4,2以及10 mg/kg时,在4周后细菌负荷均减少90%以上[38]。此外,特雷贝克在小鼠急性毒性模型中显示出良好的药代动力学性质以及安全性。
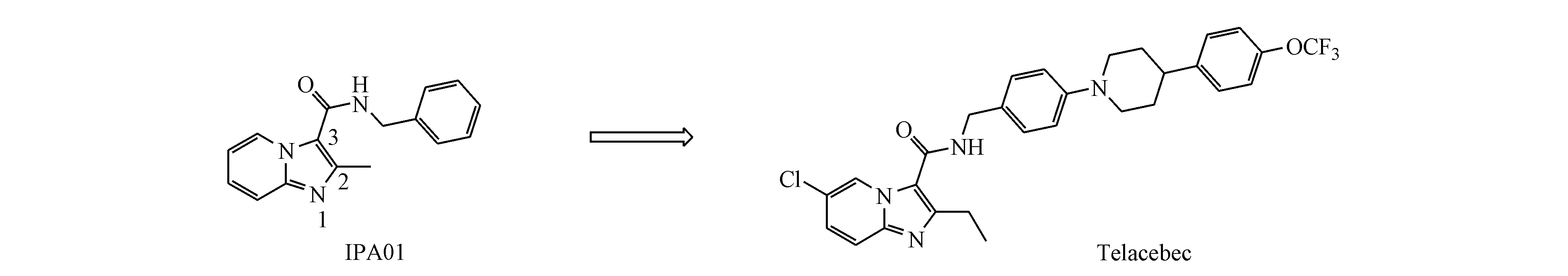
图6特雷贝克(telacebec)的发现
3 靶向蛋白质合成
3.1 舒特唑利德(sutezolid,PNU-100480)
舒特唑利德是利奈唑胺的类似物,目前处于临床Ⅱ期研究中,作用机制是通过阻断翻译来抑制蛋白质的合成。其发现起源于杜邦公司发现的唑烷酮类抗菌剂DuP 721,它对Mtb有很好的活性(MIC=1.25~4 μg/mL),但安全性研究中在100 mg/kg 的剂量下其表现出对大鼠致命的毒性。Upjohn公司发现引入哌嗪部分可以得到活性保持的化合物5,而将哌嗪中的氮原子替换为不同价态的硫得到衍生物6(图7),其中硫吗啉类化合物舒特唑利德的抗结核活性较好(MIC≤0.125 μg/mL)[39]。Williams等[40]认为在小鼠模型中它比利奈唑胺更有效。其与现有的抗结核药物没有交叉耐药性,联合使用时可以显著增加杀菌活性,表明其具有缩短结核化疗时间的潜力[41]。在Ⅰ期临床试验研究中发现舒特唑利德高效安全,耐受性良好,剂量高达1 200 mg/d,最长使用14 d,或600 mg,每日两次,最长使用28 d[42-43]。
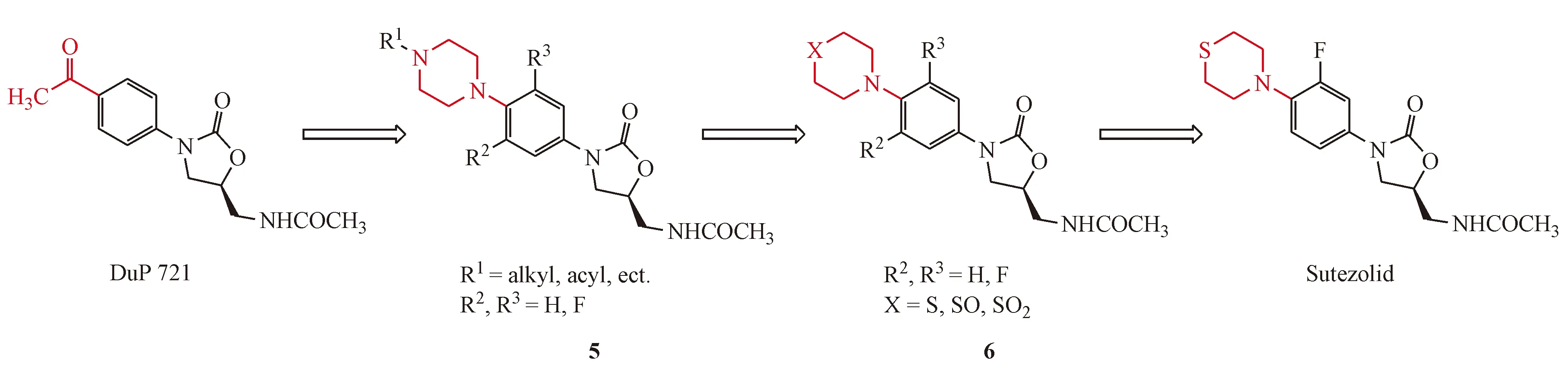
图7舒特唑利德(sutezolid)的发现
3.2 TBI-223
3.3 GSK 3036656(GSK 656,GSK 070)
GSK 3036656属于氧杂硼杂环类化合物,目前处于临床Ⅱ期研究。GSK3036656通过靶向LeuRS抑制蛋白质的合成,LeuRS属于Ⅰ类氨酰-tRNA合成酶(AARS),而AARS是所有细胞中蛋白质合成所必需的酶家族。
首先针对Mtb H37Rv菌株筛选了含20种苯并氧杂硼酸酯的化合物库,得到了MIC为1 μg/mL的AN3016和1.8 μg/mL的AN3017,接着将3位氨基甲基和7位乙氧基同时引入结构中得到化合物7(MIC =0.26 μg/mL),再在4位引入卤原子后得到活性更好的化合物8(MIC=0.02 μg/mL)。但化合物8因抑制哺乳动物细胞质LeuRS而存在潜在毒性问题,进一步优化其选择性得到了GSK 3036656(图8)。它对LeuRS的IC50为0.20 μmol/L,对Mtb H37Rv菌株的MIC为0.08 μmol/L[44]。Tenero等[45]首次在人体对GSK 3036656进行了评估,结果显示,单次给药和重复给药都有较高的安全性、良好的耐受性以及较好的药代动力学性质。
图8GSK 3036656的发现
4 结语与展望
除了靶向Mtb之外,近年来研究人员越来越关注宿主导向的治疗药物,这是因为结核病的肺损伤是普遍性的、永久性的。临床结果表明,患有结核病的患者失去了三分之一的1 s用力呼气量,治疗结束后仅恢复了一小部分[46];另一项长期研究报道,在结核病被治愈16年内,71名患者中有48名(68%)患者肺功能异常[47]。而宿主导向的治疗药物可以通过促进自噬、抗微生物肽的产生、巨噬细胞效应以及减轻肺部炎症和基质破环来缩短治疗时间、预防耐药和减少肺损伤[48]。
耐药问题给结核病治疗带来了巨大挑战,即使是上市不久的新药也有耐药现象的报道。尽管目前多个药物(表1)以及联合用药方案正处于不同阶段的临床研究中,但是考虑到新药研发的难度较大,未来仍需要继续加大投入,寻找新靶点并开发新作用机制药物来改善治疗方案,以期缩短治疗时间,提高治愈率和避免肺损伤。
表1临床上的抗结核新药

药 物结构类别 靶点/作用机制临床试验阶段 参考文献德拉马尼硝基咪唑抑制分枝菌酸合成EMA上市[6-10]普托马尼硝基咪唑抑制蛋白质和细胞壁脂质的合成US FDA上市[11-18]SQ109乙二胺MmpL3Phase II[19-22]BTZ-043苯并噻嗪酮DprE1Phase I[23-26]马考齐酮苯并噻嗪酮DprE1Phase II[27-30]OPC-167832二氢喹诺酮衍生物DprE1Phase I[31]TBA-7371氮杂吲哚DprE1Phase I[32]贝达喹啉二芳基喹啉类ATP合酶US FDA上市[35-36]特雷贝克咪唑并吡啶酰胺QcrBPhase II[37-38]舒特唑利德唑烷酮抑制蛋白质合成Phase II[39-43]TBI-223唑烷酮抑制蛋白质合成Phase I[31]GSK 3036656氧杂硼杂环LeuRSPhase II[44-45]
猜你喜欢
纺织学报(2022年12期)2023-01-06
中国防痨杂志(2022年7期)2022-11-25
昆明医科大学学报(2022年4期)2022-05-23
中国防痨杂志(2022年3期)2022-03-11
纺织检测与标准(2021年1期)2021-12-05
食品安全导刊(2021年20期)2021-08-30
昆明医科大学学报(2021年4期)2021-07-23
中华养生保健(2020年10期)2021-01-18
中华养生保健(2020年7期)2020-11-16
生物工程学报(2020年1期)2020-03-12
