
幽门螺杆菌毒力蛋白cagA基因敲除突变株构建及鉴定*
2020-05-06 01:35:12李抄陈定宇张晓怡赵艳王琴容周建奖谢渊
贵州医科大学学报 2020年2期
李抄,陈定宇,张晓怡,赵艳,王琴容,周建奖,谢渊
(贵州医科大学 地方病与少数民族性疾病教育部重点实验室,贵州省医学分子生物学重点实验室,贵州医科大学 分子生物学重点实验室,贵州 贵阳 550004)
胃癌是世界上最常见的恶性肿瘤。据2018年全球流行病学资料显示,全球胃癌的发病率居恶性肿瘤的第6位,死亡率居第5位;在中国,胃癌的发病率和死亡率分别占全部肿瘤的第4位和第2位[1-2]。胃癌的发生发展涉及多个方面,主要包括宿主遗传学、环境因素和幽门螺杆菌(Helicobacterpylori,Hp)感染[3]。Hp是一种螺旋状革兰阴性微需氧菌,它定殖在人类的胃黏膜上皮,是引起胃炎、消化性溃疡、胃癌的主要病因。Hp的发现加深了人类对慢性感染、炎症和癌症之间关系的认知,1994年世界卫生组织将Hp列为 I 类致癌因子[4-5]。全世界有近半数人感染Hp,但仅少数会发展成胃癌,究其原因,除宿主与环境因素外,HpcagA毒力表达是导致其感染后不同临床结局的可能原因[6]。细胞毒素相关基因A蛋白(cytotoxin associated gene A,cagA )是由HpcagA致病岛编码的一种120~145 kDa的蛋白质,通过IV型分泌系统(type Ⅳ secretion system,TFSS)注入胃上皮细胞,参与胃癌的发生发展,是目前所知的唯一被Hp注入胃上皮细胞并能模拟细胞内蛋白发挥作用的“癌蛋白”(oncoprotein),但其致癌机制还不完全清楚[7-9]。基因打靶(gene targeting)是20世纪80年代发展起来的一项重要的分子生物学技术,是利用基因转移方法将外源DNA序列导入靶细胞后,通过外源DNA 序列与靶细胞内染色体上同源DNA序列间的重组,将外源DNA定点整合入靶细胞基因组上某一确定的位点,或对某一预先确定的靶位点进行定点突变,从而改变细胞遗传特性的方法[10-11]。目前基因打靶技术已应用在改造生物、培育新的生物品种,研究基因结构与功能、表达与调控,研究细胞生活周期调控机制,遗传病的基因治疗等多方面。为进一步明确cagA在Hp致胃癌发生发展中的作用,本实验运用基因打靶技术构建敲除cagA基因的Hp突变株,为后续研究cagA致病机制奠定实验基础。
1 材料与方法
1.1 材料、主要试剂及仪器
1.1.1材料 pACYC184质粒、pUCmT载体及细菌DNA提取试剂盒(上海生工生物公司),幽门螺杆菌HP1004及胃癌原代细胞(贵州医科大学分子生物重点实验室保存)。
1.1.2主要试剂和仪器 双抗(美国 HyClone公司),改良型RPMI-1640 培养基和胎牛血清(美国Gibco 公司),氯霉素(chloramphenicol,Cm,中国 Beyotime公司),哥伦比亚血琼脂培养基(英国Oxoid公司),Taq DNA聚合酶和T4 DNA ligase(日本TAKARA公司),anti-cagAantibody和anti-GAPDHantibody(美国CST公司);电转化仪Electroporator 2510(中国 Eppendorf),全制动化学发光成像分析仪(中国 上海天能科技公司);Primer5.0设计引物合成于上海生工,见表1。
1.2 方法
1.2.1cagA基因的上、下游同源重组臂扩增 高保真PCR酶从Hp基因组上扩增cagA基因的上、下游同源重组臂,反应体系为50 μL[Hp基因组DNA 0.5 μL、10×pfu buffer 5 μL、dNTP(2.5×10-2mol/L) 0.5 μL、cagA-5F/3F及cagA-5R/3R (5×10-6mol/L)各0.5 μL,pfu DNA polymerase(5×10-3U/L) 0.5 μL,DMSO(5%) 2.5 μL,ddH2O补足体积];聚合酶链式反应(polymerase chain reaction,PCR)循环程序为95 ℃、5 min,95 ℃、30 s,60 ℃、30 s,72 ℃、60 s,5个循环,72 ℃延伸7 min。

表1 引物序列Tab.1 Primer sequence
1.2.2Cm的表达框序列扩增 从pACYC184质粒上扩增Cm的表达框序列,Cm抗性基因的扩增反应体系为50 μL:pACYC184质粒DNA 0.5 μL,10×pfu buffer 5 μL,dNTP(2.5×10-2mol/L) 0.5 μL,cagA-Cm-F和cagA-Cm-R (5×10-6mol/L)各0.5 μL,pfu DNA polymerase(5×10-6U/L) 0.5 μL,ddH2O 补足;PCR循环程序同方法1.2.1。
1.2.3融合PCR构建打靶片段cagA基因上、下游同源重组臂经设计与Cm抗性基因序列有部分重叠,可通过PCR直接融合,获得全长的打靶片段。反应总体系100 μL:上、下游同源重组臂PCR产物、Cm抗性基因PCR产物、5%DMSO各5 μL,10×pfu buffer 10 μL、dNTP(2.5×10-2mol/L)、cagA-5F、cagA-3R (5×10-6mol/L)及pfu DNA polymerase(5×10-6U/L)各1 μL,dH2O补足;PCR循环程序同方法1.2.1,循环数为25次。
1.2.4打靶载体(pUCmT-ΔcagA::Cm)的构建 在打靶片段PCR产物中直接加入Taq DNA聚合酶(5×10-6U/L),于72 ℃反应30 min电泳并切胶纯化后与然后与50 mg/L pUCmT载体进行连接反应,反应体系10 μL:pUCmT(5×107ng/L) 2 μL,打靶片段(5×107ng/L) 6 μL,10×T4 buffer 1 μL,T4 DNA ligase(5×10-6U/L) 1 μL;16 ℃反应过夜。
1.2.5打靶载体的电转化 取4 mL液体培养(布氏肉汤加10%胎牛血清)的Hp分装于2 mL无菌Ep管中,4 ℃、6 000 r/min离心5 min收集菌体;等体积无菌去离子水轻柔吹打洗涤,4 ℃、6 000 r/min离心5 min收集菌体,相同方法重复洗涤1次;用100 μL无菌10%甘油轻柔悬浮菌体,取40 μL分装在预冷的0.5 mL无菌Ep管中即可用作电转化细胞。取500 ng预冷的打靶质粒加入40 μL新鲜制备的电转化细胞,4 ℃放置2 min,转移入2 mm Eppendorf电击转化杯,迅速放入电转化仪,2 500 V电压下电击转化;加入Hp培养液1 mL悬浮菌体,转入50 mL无菌尖底离心管,加入液体培养基5 mL,2.5 L厌氧罐内37 ℃、100 r/min震荡培养过夜。
1.2.6cagA基因敲除菌株Hp/ΔcagA::Cm的筛选 吸取500 μL打靶载体电转化后的细菌培养液转接5 mL液体培养液(含Cm 2×10-2μg/L),于上述同样培养条件继续培养3~4 d,直至培养液浑浊;取微量浑浊培养液划线接种固体培养基(Karmali培养基,加10%脱纤维绵羊血和2×10-2μg/L Cm),于微需氧厌氧罐内培养至单克隆形成。
1.2.7cagA基因敲除菌株Hp/ΔcagA::Cm的PCR鉴定 随机挑选若干阳性克隆,分别接种3 mL布氏肉汤(含10%胎牛血清),厌氧罐条件下37 ℃、100 r/min培养至菌体明显浑浊,取少量菌液进行PCR扩增;用cagA外侧引物扩增时,如cagA基因未被替换,PCR产物长度为5 014 bp;而打靶载体替换成功时,产物长度则缩短为2 150 bp;反应体系50 μL:基因组DNA 0.5 μL,10×TaqPlus buffer 5 μL,dNTP(2.5×10-2mol/L) 0.5 μL,cagA-outF及cagA-outR(5×10-6mol/L)各0.5 μL,5% DMSO 2.5 μL,Taq Plus DNA polymerase(5×10-6U/L) 0.5 μL及ddH2O 补足;PCR循环程序同方法1.2.1,循环数为35次,1%琼脂糖凝胶电泳检测结果。
1.2.8原代胃癌细胞培养及Hp/ΔcagA::Cm、Hp/cagA感染原代胃癌细胞 按1.0×106/孔细胞数量接种于含DMEM完全培养基(10% FBS)6孔板中,37 ℃的 5%CO2孵育箱常规培养,感染前将6孔板中换成无双抗含Gibco血清培养基;Hp/ΔcagA::Cm和Hp/cagA培养3 d,感染复数(mutiplicity of infection,MOI) 30 ∶1的比例分别感染原代胃癌细胞,24 h后倒置荧光显微镜下观察细胞形态,并分别收集各组细胞,血球计数板计数。
1.2.9Western blot检测原代胃癌细胞内cagA存在 按全蛋白提取试剂盒进行细胞全蛋白提取,按BCA蛋白含量检测试剂盒进行蛋白定量,10%PAGE分离蛋白,上样蛋白体积20 μL,电泳,湿转PVDF膜,5% 脱脂牛奶封闭,4 ℃孵育cagA一抗(1 ∶1 000/4 000)过夜,二抗(1 ∶10 000)室温孵育2 h,TBST清洗3次,按化学发光底物试剂盒进行反应,利用荧光凝胶成像系统曝光、显影及分析。
1.3 统计学分析
使用SPSS 17.0软件进行统计学分析,Graphpad Prism 7.0软件绘图,组间比较使用独立样本t检验,以P<0.05为差异有统计学意义。
2 结果
2.1 融合PCR构建打靶片段鉴定
cagA基因上、下游同源重组臂经设计与Cm抗性基因序列有部分重叠,可通过PCR直接融合,获得全长的打靶片段,长度为1 794 bp,见图1。
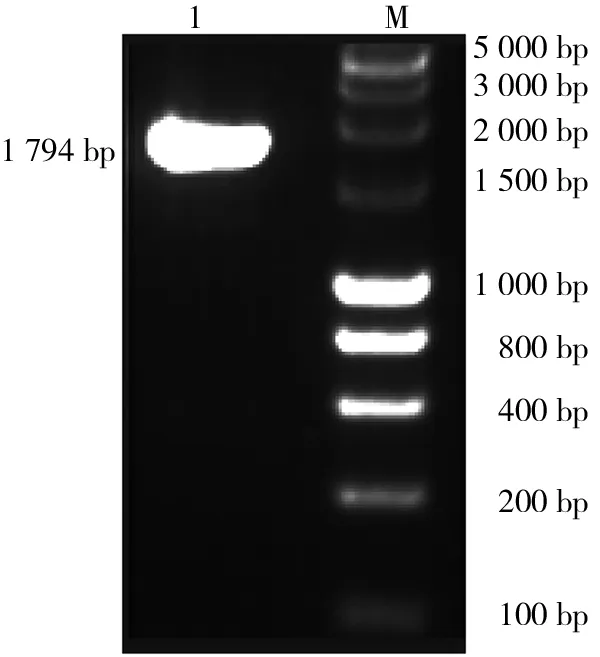
注:1为打靶片段,M为标准分子量参照(DNA Maker)。图1 打靶片段融合PCR结果Fig.1 Target fragment fusion PCR map
2.2 Hp/ΔcagA::Cm突变株PCR鉴定
PCR扩增鉴定结果显示,Hp/ΔcagA::Cm阳性克隆扩增产物为2 150 bp,野生型Hp/cagA菌株扩增产物为5 014 bp,表明成功获得Hp/ΔcagA::Cm敲出突变菌株,见图2。

注:1为野生Hp/cagA菌株,2为 Hp/ΔcagA::Cm突变株,M 为DNA分子量标准。图2 Hp/ΔcagA::Cm突变株PCR鉴定结果Fig.2 Hp/ΔcagA::Cm mutant PCR identification map
2.3 Hp/ΔcagA::Cm和Hp/cagA感染的原代胃癌细胞形态
Hp/ΔcagA::Cm和Hp/cagA分别感染3株胃癌原代细胞,24 h后荧光倒置显微镜下观察细胞形态改变,可见感染野生型Hp/cagA的原代胃癌细胞发生明显的细胞形态改变,由钝圆变为长梭形、纺锤形、不规则形,细胞死亡数量多,感染Hp/ΔcagA::Cm突变株的细胞形态改变较小,死亡细胞数量少,且差异有统计学意义(P<0.05或P<0.01),见图3。
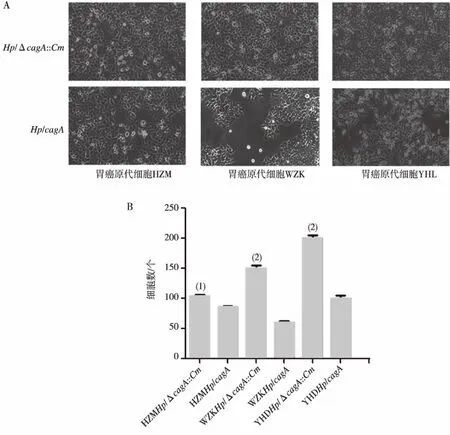
注:A为Hp/ΔcagA::Cm和Hp/cagA感染3株胃癌原代细胞,B为细胞数量的直条图;(1)与WZKHp/cagA,YHDHp/cagA组比较,P<0.01;(2)与HZMHp/cagA组比较,P<0.05。图3 Hp/ΔcagA::Cm和Hp/cagA感染细胞形态改变 (100×)Fig.3 Hp/ΔcagA::Cm and Hp/cagA infected cell morphology changes (100×)
2.4 原代胃癌细胞内cagA蛋白表达
Western blot检测cagA蛋白(分子量135 kDa)表达情况,结果表明野生型Hp/cagA菌株及其感染3株原代细胞内cagA蛋白表达水平较Hp/ΔcagA::Cm突变型菌株及其感染3株原代细胞明显降低,差异有统计学意义(P<0.01),见图4。

注:A为Western blot检测cagA蛋白条带,B为cagA蛋白表达量的条图;(1)与HZMHp/ΔcagA::Cm组比较,P<0.01;(2)与WZKHp/ΔcagA::Cm组比较,P<0.01;(3)与YHDHp/ΔcagA::Cm组比较,P<0.01;(4)与Hp/ΔcagA::Cm组比较,P<0.01。图4 细菌及其感染细胞内cagA 表达情况Fig.4 CagA expression in bacteria and infected cells
3 讨论
全球约50%人口存在Hp感染,多数感染患者为无症状胃炎,少数发展为萎缩性胃炎甚至胃癌[12]。cagA是HP感染引起炎症反应的重要效应蛋白,被Hp注入胃上皮细胞的cagA发生磷酸化,磷酸化的cagA和Scr同源区2结合激活Scr同源区的磷酸化酶活性,从而引起瀑布式的级联反应;通过干扰细胞信息传导通路引起组织严重的炎症损伤,参与胃癌的形成过程[13]。cagA可引起胃癌细胞的增殖和凋亡紊乱[14]。临床检出的Hp包括cagA阳性和cagA阴性2种类型;cagA阳性Hp感染患者发生胃癌的风险高于cagA阴性者[15]。现有研究认为cagA与Hp引起疾病的严重程度相关;Hp/cagA+菌株可引起更严重的炎症反应,更容易引起胃萎缩和胃腺癌的发生,是Hp/cagA-菌株的2.0~28.4倍[16-17];因此cagA在胃癌的发生发展过程中发挥重要作用。为了更好地研究Hp中cagA蛋白的致病功能,通过基因打靶的同源重组原理构建了HpΔcagA基因缺失突变株,为系统研究cagA基因功能创造了条件,也为进一步阐明Hp致病机制奠定基础。
基因打靶是分析基因功能的重要技术,用于定位功能基因,研究模式生物和一些重要微生物的全基因组功能,被广泛应用于生命科学与医学的研究领域[18]。基因打靶技术以同源重组为理论基础,同源重组机制在原核和真核生物中普遍存在,是保证物种遗传信息稳定性和多样性的一种重要机制[19]。有研究发现,新的遗传物质可以通过同源重组引人生物细胞的基因组,井对基因进行特异性修饰和改造[20-22];也有研究报道,构建出了cagA全基因缺失的Hpylori突变株[23-24],本研究利用高保真PCR酶从Hp基因组上扩增cagA基因的上、下游同源重组手臂;从pACYC184质粒上扩增Cm表达框序列。通过融合PCR技术获得cagA基因敲除用的打靶片段(上游同源手臂-氯霉素抗性基因-下游同源手臂),将其克隆入通用载体pUCmT,获得打靶载体pUCmT-ΔcagA::Cm。然后通过电转化,将pUCmT-ΔcagA::Cm质粒直接转入Hp,在抗生素压力的帮助下,打靶片段和菌体基因组发生同源重组,通过氯霉素抗性筛选得到带有抗性标记的重组菌,随后因突变载体不能在Hp1004菌体内生长复制而丢失。
检测突变株Hp/ΔcagA::Cm是否还存cagA基因或蛋白,是最后认证cagA基因缺失突变株构建成功与否的关键。本研究对Hp/ΔcagA::Cm突变型菌株和野生型Hp/cagA菌株的基因组进行了PCR鉴定,用cagA外侧引物扩增时,野生株可以扩出完整的cagA基因和非编码序列的长约5 014 bp,而cagA缺失突变株cagA基因被打靶载体替换,产物长度则缩短为2 150 bp。另外本研究用Hp/ΔcagA::Cm突变型菌株和野生型Hp/cagA菌株感染原代胃癌细胞,Western blot实验均能从野生型Hp/cagA菌株及其感染细胞检出cagA蛋白,而Hp/ΔcagA::Cm突变型菌株及其感染细胞内均不能检出cagA蛋白,从而在蛋白水平上证实了上述实验结果,表明本次cagA缺失突变株Hp/ΔcagA::Cm构建成功。同时本研究还发现感染野生型Hp/cagA的原代胃癌细胞发生明显的细胞形态改变,由钝圆变为长梭形、纺锤形及不规则形,细胞死亡数量多,而感染Hp/ΔcagA::Cm突变株的细胞形态改变较小、死亡细胞数量少;这进一步证实cagA被Hp注入细胞后对细胞有一定的毒性作用。
综上所述,本实验成功构建了Hp/ΔcagA::Cm突变株,并在感染细胞中成功验证,且证实cagA蛋白对细胞有一定的毒性作用,为进一步研究Hp中cagA基因的功能,阐明其在Hp致病中的地位及作用奠定了实验基础。
猜你喜欢
——紫 苏
河南农业(2024年1期)2024-01-19 01:56:54
华人时刊(2023年1期)2023-03-14 06:43:36
汉字汉语研究(2021年2期)2021-08-30 08:58:46
今日农业(2021年11期)2021-08-13 08:53:24
解放军医学院学报(2020年12期)2020-03-29 05:11:32
心肺血管病杂志(2019年9期)2019-12-09 08:34:02
中成药(2018年9期)2018-10-09 07:18:32
河北书画研究(2016年3期)2016-04-28 08:55:35
中华皮肤科杂志(2014年3期)2014-12-19 12:54:50
遗传(2014年3期)2014-02-28 20:58:49
