
VPS13A基因突变致舞蹈病-棘红细胞增多症的诊断及家系研究*
2020-05-04 07:58:50姚念廷郑乾冯占辉尹建红徐子茜焦玲张春林
贵州医科大学学报 2020年3期
姚念廷,郑乾,冯占辉,尹建红,徐子茜,焦玲*,张春林
(1.贵州医科大学附院 神经内科,贵州 贵阳 550004;2.贵州医科大学 基础医学院,贵州 贵阳 550004)
舞蹈病—神经棘红细胞增多症(chorea-acanthocytosis,ChAc)是一种临床罕见的以舞蹈样动作和外周血棘红细胞增多为典型特征的神经系统退行性疾病,临床表现主要为口、舌、面不自主运动、进行性舞蹈样运动障碍、精神症状、认知功能下降以及其他多系统损害[1]。该病临床表现形式多样,可累及运动、神经、精神以及内分泌等全身多个系统[2-6],与麦克劳德综合征(mcLeod neuroacantho cytosis syndrome,MLS)、类亨廷顿病2型(huntington’s disease-like 2,HDL2)、泛酸激酶相关的神经退行性(pantothenate kinase—associated neurodegeneration,PKAN)等疾病的临床症状、实验室检查及影像学表现上存在较多的重叠,其临床诊断比较困难[7]。目前ChAc还没有具体的诊断标准,但ChAc的早期诊断至关重要,外周血棘红细胞是该病的重要特征性的诊断依据,而分子生物学及基因检测是ChAc诊断的金标准[8-10]。ChAc是常染色体隐性遗传疾病[5,7,11],液泡蛋白分类同源物13A(vacuolar protein sorting homolog 13A,VPS13A)基因是目前唯一与ChAc相关的致病基因[10,12],目前已报道VPS13A基因突变形式多样化,包括错义突变、无义突变、移码突变、缺失或插入突变等,这些不同类型的突变增加了ChAc遗传诊断复杂性[5,13]。因ChAc为十分罕见的遗传病,故本研究通过收集的1例疑似ChAc病例及其家系的临床资料,同时利用扫描电镜检测外周血棘红细胞及全外显子目标序列捕获2代测序技术鉴定其基因突变点,探讨ChAc的诊断方法,为ChAc的精准诊断提供可行合理的策略,以期避免误诊或漏诊。
1 对象和方法
1.1研究对象、主要试剂和仪器
1.1.1研究对象 收集某院1例临床表现疑似ChAc先证者及其家系资料,其中先证者符合以下ChAc诊断标准[5],即肢体舞蹈样动作,口周和(或)口下颌肌张力障碍及进食性肌张力障碍,口唇自噬动作,颅脑MRI可见尾状核头萎缩,血生化提示肌酸激酶(creatine kinase,CK)、肌酸激酶同工酶(creatine kinase isoenzymes,CK-MB)、乳酸脱氢酶(lactate dehydrogenase,LDH)、谷丙转氨酶(alanine aminotransferase,ALT)及谷草转氨酶(aspartate aminotransferase,AST)均升高。先证者(Ⅱ3)29岁、男性、汉族、既往体健,先证者父亲(Ⅰ1)57岁、汉族,先证者母亲(Ⅰ2)55岁、汉族,先证者哥哥(Ⅱ1)35岁时去世、汉族,先证者姐姐(Ⅱ2)32岁、汉族。
1.1.2主要试剂和仪器 2.5%戊二醛固定液(上海釜诚生物科技有限公司),0.1 mol/L PBS缓冲液(北京百奥莱博科技有限公司),50%、70%、80%、90%和100%酒精(北京柏奥易思生物科技有限公司),标准文库构建试剂盒(北京迈基诺基因科技股份有限公司),DNA 提取试剂盒(德国Qiagen公司),Illumina标准测序试剂盒(美国NEB公司);XL-30扫描电镜(荷兰PHILIPS公司),BAL-TEC SCD离子溅射仪(瑞士BAL-TEC公司),Covaris S2超声仪(美国Covaris公司),ABI 3730XL测序仪(美国应用生物系统公司),Nanodrop 2000 样本定量检测仪(美国Thermo Fisher科技有限公司)。
1.2方法
1.2.1先证者临床信息 收集先证者基本临床资料、实验室和影像学结果,询问并记录家系情况并绘制家系图。
1.2.2电镜检查 提取先证者保留外周静脉血1~2滴加入5 mL新鲜配制的质量分数2.5%戊二醛固定液中,轻轻振荡后静置于4 ℃冰箱0.5 h,取适量已固定好血样置于1.5 mL离心管,1 500 r/min低速离心15 min去上清,0.1 mol/L PBS缓冲液漂洗3次、5 min/次,体积分数50%、70%、80%、90%及100%酒精(2次)脱水,加入适量无水乙醇混匀,滴于洁净盖玻片上,室温自然干燥;用双面胶带将干燥后的盖玻片粘贴在样品台上,利用BAL-TEC SCD离子溅射仪喷金,利用PHILIPS XL-30扫描电镜观察棘红细胞。
1.2.3筛查致病基因 (1)DNA提取、基因组文库构建、基因的捕获及高通量测序:提取先证者保留外周静脉血2 mL,使用基因组DNA提取试剂盒对样本DNA进行核酸提取;取先证者的DNA 3 μg稀释,采用Covaris S2超声仪进行超声片段化(大小150 bp);用标准文库构建试剂盒制备全基因组文库,用Nanodrop 2000样本定量检测仪进行质控;采用GenCap技术(北京迈基诺基因科技有限责任公司)捕获目的基因,用Illumina NextSeq 500进行测序。(2)数据筛选和生物信息学分析:目标区域测序后,去除测序数据中的接头和低质量数据 (质量值≤20) ,运用BWA软件比对参考基因组,用GATK软件对各个样本的比对数据进行多态性位点的检测,对单核苷酸多态性( single nucleo- tide polymorphisms,SNPs)和插入缺失突变 (InDels)等数据进行统计和分析;同时进行测序深度、均一性、探针特异性等数据的统计分析;查找SNPs及In-Dels 在千人基因组(http://www.1000genomes.org/)及dbSNP132(http://www.ncbi.nlm.nih.gov/pro-jects/SNP/)数据库中频率,利用PolyPhen-2软件(http://genetics.bwh.harvard.edu/pph2/)进行SNPs及InDels 致病性分析。(3)Sanger测序验证:根据需要测序的DNA片段合成引物,用聚合酶链反应(polymerase chain reaction,PCR)法进行扩增,用ABI 3730xl 测序仪以Sanger测序法进行测序,测序结果与目标区域捕获测序后的结果进行比对。
2 结果
2.1先证者的临床资料和家系图
先证者(Ⅱ3),29岁男性,因“进行性口面部、四肢不自主运动2+年,加重半年”于2018年11月22日入院。2+年前先证者无明显诱因逐渐出现伸舌、磨牙、扮鬼脸、头部摆动及四肢舞蹈等不自主动作,伴反复下唇咬伤、溃疡,近半年症状逐渐加重,伴吞咽困难,无法进食。神经系统查体:言语欠流畅、吐词含混,张口稍受限,口唇及舌尖有多处咬伤、溃疡;眼周、口周、颈部、四肢可见不自主运动;颈部肌张力增高,双上肢肌力减退,四肢腱反射减弱,四肢共济运动欠稳准。辅助检查CK、CK-MB、LDH、ALT及AST均升高。神经传导检查回示感觉性周围神经损害,颅脑MRI可见尾状核头轻度萎缩。先证者哥哥(Ⅱ1),35岁去世,生前症状与先证者一致;先证者父亲(Ⅰ1),先证者母亲(Ⅰ2),近亲结婚,体健;先证者姐姐(Ⅱ2),体健。见图1、图2。
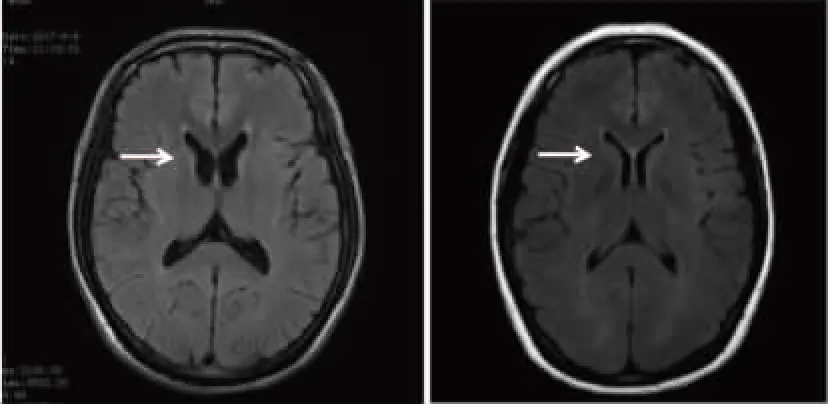
先证者 正常人注:箭头所指为尾状核头。图1 先证者与正常人的颅脑MRI图像对比Fig.1 Comparison of MRI images between the proband's and normal human's brain

图2 先证者家系图Fig.2 Family map of the proband
2.2先证者外周血红细胞的扫描电镜
先证者外周血扫描电镜结果可见典型棘红细胞(正常人外周血无棘红细胞)。见图3。

注:A为放大倍数6 000×,B为放大倍数10 000×;箭头所示为典型棘红细胞。图3 先证者外周血红细胞扫描电镜结果Fig.3 Scanning electron micrograph of peripheral blood red blood cells in the proband
2.3致病基因筛查
通过高通量全外显子测序和突变位点生物信息学分析结果表明,先证者检测出VPS13A基因且存在c.9403C>T纯合突变(基因型为TT纯合子),引起chorein蛋白p.R3135X突变,为无义突变;SIFT数据库预测结果为“damaging”,PolyPhen-2数据库预测结果为“probably damaging”。先证者的父亲和母亲均为c.9403C>T杂合突变。见图4。

图4 先证者及其父母VPS13A基因核苷酸c.9403C>T突变位点测序结果Fig.4 Sequencing of nucleotide mutations c.9403C>T in the VPS13A gene of proband and his parents
3 讨论
神经—棘红细胞增多症 (neuroacanthocytosis,NA) 是一类罕见的神经遗传性疾病,具有低患病率和高临床变异性的特点[9]。NA可分为ChAc、MLS、HDL2和PKAN4种亚型[14-15],其中以ChAc最多见[16]。ChAc常见于20~30岁人群,临床表现包括[14,17-18]:(1)运动过多型运动障碍,如肢体舞蹈样动作,口腔、面、舌不自主运动,进食性肌张力障碍、扮鬼脸、怪异表情等为该病最常见症状;(2)口唇自噬动作,表现为不自主唇舌咬伤,伴口腔溃疡,该症状对该病具有特异性诊断价值[5];(3)磨牙征,表现为不自主牙齿咀嚼行为;(4)帕金森病样表现,如运动迟缓、静止性震颤、肌张力增高等;(5)癫痫发作,其中以强直—阵挛发作多见;(6)认知障碍,以注意力和执行功能为主;(7)神经精神症状,表现为淡漠、抑郁及焦虑等;(8)周围神经损害症状,表现为麻木、无力、肌萎缩、腱反射减弱或消失。ChAc在影像学检查上可见尾状核(尤其是尾状核头)和豆状核萎缩、低代谢、低灌注,相应可见侧脑室前脚扩大[17];临床生化检查中可见肌酶、转氨酶增高,其中肌酸激酶增高更具有特异性[14]。本例患者主要表现为口面部不自主动作、口唇自噬及四肢舞蹈样动作,且相关辅助检查均符合该疾病的特异表现,故根据临床资料高度考虑ChAc。此外,有研究认为,此类患者外周血涂片或者扫描电镜检测到典型棘红细胞,且典型棘红细胞增多大于3%对于ChAc具有重要诊断价值[1,5]。本例患者外周血扫描电镜检测到了典型棘红细胞,比例约25%,故进一步支持ChAc的诊断。但外周血棘红细胞在MLS中同样也较为常见[1,12],故不易鉴别。
根据以上临床资料及扫描电镜结果,目前基本可以明确为ChAc,但ChAc与NA其他3种亚型在临床特征、实验室检查及影像学表现上存在交叉性,鉴别诊断存在较大困难[7]。故本研究进一步行基因检测。本例先证者检测到VPS13A基因的c.9403C>T点纯合突变(基因型为TT纯合子),可导致chorein蛋白的p.R3135X突变,且系低频变异;SIFT数据库预测结果为"damaging",PolyPhen-2数据库预测结果为"probably damaging",按照ACMG发布的最新基因变异解读标准和指南,考虑为可能致病(likely pathogenic)。同时,结合先证者家系资料,其兄生前具有与先证者一致的临床表现,均表现头面部及肢体的舞蹈样动作。而先证者父母均为杂合突变(基因型为CT杂合子),并且临床表现无舞蹈症状。因此,说明VPS13A(c.9403C>T)点突变在本研究ChAc遗传家系中符合常染色体隐性遗传方式。已有研究表明ChAc以常染色体隐性遗传为主,定位于9q21的VPS13A基因是目前公认已知的致病基因,翻译产物是chorein蛋白[19]。红细胞是由膜脂质、膜蛋白及血影蛋白—肌动蛋白骨架协同维持[20],而chorein蛋白参与了肌动蛋白的聚合反应,其功能障碍可能导致细胞膜破裂和异常的红细胞形状[19]。故VPS13A基因突变导致chorein蛋白的改变对该疾病有重要的诊断价值[12]。VPS13A基因突变形式多样,不同VPS13A基因突变可引起chorein蛋白功能性缺如或合成减少,最终导致ChAc的发生与发展[5,21]。目前,VPS13A基因测序和chorein蛋白定量检测被认为是ChAc的确诊性诊断标准[5]。因此,本文结果与已经报道的ChAc遗传家系VPS13A基因遗传方式相符合[5,16]。在鉴别诊断方面,MLS是一种罕见的X连锁疾病,由XK基因突变引起[22];HDL2是一种常染色体显性遗传性疾病,由位于6q24-3染色体的JPH3基因内的CTG/CAG三核苷酸重复扩增引起[23-24];PKAN是一种染色体20p13上编码泛酸激酶2的PANK2基因突变引起的常染色体隐性遗传疾病[25-26]。因此,通过基因检测不仅可以准确诊断ChAc,并且在鉴别诊断方面亦有重要的意义。
综上所述,虽然结合临床表现及扫描电镜检查可以高度怀疑ChAc,但全外显子目标序列捕获2代测序技术可准确检测VPS13A基因突变,并结合遗传家系特点,可作为ChAc首选确诊手段。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
智富时代(2018年7期)2018-09-03 03:47:26
电线电缆(2017年4期)2017-07-25 07:49:48
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
中国石油大学学报(自然科学版)(2015年2期)2015-11-10 06:07:37
