
苯系物Cu3金属配合物的结构与性质的理论研究
2020-04-28 04:19冷艳丽张榕芳慕红梅张建辉
原子与分子物理学报 2020年1期
刘 婧, 冷艳丽, 张榕芳, 慕红梅,张建辉,
(1.甘肃农业职业技术学院 食品化工系, 兰州 730020; 2.贵州民族大学 化学工程学院, 贵阳550025; 3.兰州资源环境职业技术学院 环境与化工系, 兰州730021 )
1 引 言
伴随着全球工业化水平的提高和经济的快速发展,大气污染、水污染和噪音污染已严重危害地球和人类生存环境,尤其大气污染的不可见性和强扩散性对生物造成长期性的危害.大气污染物成分复杂,除了悬浮在大气中的颗粒污染物,还有混合在空气中的气态污染物[1]. 其中,挥发性有机化合物(Volatile Organic Compounds, VOCs)是一类重要的气态污染物,其通常指室温下饱和蒸气压超过 70.91 Pa 或沸点小于 260 ℃的有机物[2],挥发性有机污染物种类众多,目前已鉴别出 300 余种,其分类遍布卤代烃、脂肪烃、芳香烃、 醇、 醛、 酮、 醚、 酯及多环芳烃[3]. 挥发性有机污染物主要来自以煤、石油、天然气为燃料或原料的化工企业. 有效的降解治理该类污染物已成为控制大气质量和保护环境的必由之路. 以苯系物为代表的VOCs极易挥发,可通过各种方式进入人体,对体内各种器官都有致癌性,对人类健康造成巨大危害[4].因此,解决这类挥发性有机化合物显得十分重要和紧迫. 由于VOCs 种类较多, 不同性质不同浓度的污染物净化方式又不同,针对不同的污染物与浓度,其治理方法可分为回收法、降解法和燃烧法三大类[5-10], 其中燃烧法有直接燃烧法和催化燃烧法[11, 12].催化燃烧技术具有起燃温度低、适用范围广、处理效率高和无二次污染等优点, 被认为是最为经济有效的治理技术之一.
催化燃烧技术中常用催化剂主要有贵金属催化剂和过渡金属氧化物催化剂. 其中, 贵金属催化剂一般为Pt、Pd、Rh、Au等贵金属, 该类催化剂寿命长,起燃温度低,完全燃烧温度与起燃温度相差较小,一旦达到某个温度可使转换率直线上升,缺点是价格昂贵、资源缺乏且容易中毒, 限制了其推广应用[13,14]. 过渡金属氧化物催化剂, 如Cr、Cu、Co、Mn的氧化物, 低温活性虽然不如贵金属催化剂, 但它们价格低廉, 从结构分析主要是由于这些过渡金属中的金属离子最高占有轨道和最低空轨道是 d 轨道和 f轨道,或是 d 轨道和 f轨道的杂化,金属离子容易变价, 具有很好的应用前景[15]. 如Cu、Mn、Fe、Co、Ce等复合型催化剂表现出更高的催化剂活性和更好的稳定性[16-18].在非均相催化反应中, 过渡金属催化剂常常以不同尺寸的团簇负载于载体表面, 不同尺寸的金属簇催化活性也不相同, 由此认识团簇几何结构及其稳定性、研究反应物与催化剂之间的相互作用可揭示催化作用本质.
甲苯、二甲苯、三甲苯等苯系物都是的挥发性有机物, 了解过渡金属在它们催化燃烧中的作用, 将为复合型催化剂的制备提供理论支持. 本文将结合前人在甲苯、二甲苯、三甲苯等苯系物吸附实验中的结论[17.19-23], 以铜团簇Cun(n=1-6)中前线轨道的能级差最小, 活性最大的Cu3[24]和甲苯、二甲苯、三甲苯作用为研究对象, 从吸附能、前线轨道等角度分析各配合物结构与性质, 从微观角度了解该类配合物的作用机理, 对设计和合成催化剂提供理论指导.
2 计算方法
采用密度泛函理论中的混合密度泛函方法B3LYP[25,26], 和6-311+G(d,p)基组[27], 对铜团簇结构和甲苯、二甲苯、三甲苯配合物结构进行全参数优化, 得到各结构的键长、键角、振动频率等参数, 通过频率分析证实了结构的稳定性, 根据能量搞定确定基态的多重度, 我们还采用自然键轨道(Natural Bond Orbital, NBO)方法分析了其成键情况以及轨道间的相互作用[28-30]. 全部计算工作采用Gaussian09程序完成[31], 分子的几何构型全部由Gauss View程序从计算结构直接转换而来,计算所得结构如图一所示.
3 结果与讨论
根据研究小组先前研究苯与Cun(n=1-6) 团簇作用过程, 我们得到能级差最小的Cu3能隙差为0.014 a.u., 在Cun(n=1-6)中活性越大, 与苯分子间形成的键越牢, Cu-C键长最短且放出的能量最高[24]. 基于以上研究结果,本文将进一步运用密度泛函方法对Cu3团簇与甲苯、二甲苯、三甲苯等苯系物作用的各种异构体进行优化计算, 得到基态的最稳定结构. 进而研究它们的吸附能等性质.
3.1 Cu3(CH3C6H5)配合物
根据计算结果所示,Cu3与甲苯作用的最稳定结构为:Cu3团簇中一个Cu原子与苯环平面上甲基相邻的β、γ碳原子发生作用新生成三元环结构,Cu-C键长分别为2.206和2.214 Å,距甲基较近的键略微短一些,体系的吸附能为58.3 kJ·mol-1. 依据前线轨道理论, 各构型前线轨道的能级差Egap(Egap=ELUMO-EHOMO)越小, 活性越大, 分子间形成的键越牢, 体系稳定化程度就越大[32-35]. 计算得到甲苯的能级差为0.1818 a.u., Cu3(CH3C6H5)的能级差为0.0113 a.u., 通过比较我们很明显发现Cu3与CH3C6H5配位反应明显降低了体系的能级差, 对进一步催化燃烧将起到显著的促进作用.
3.2 Cu3(CH3)2C6H4配合物
关于Cu3与二甲苯作用体系,我们对系统研究了Cu3与二甲苯邻、间、对三种异构体作用的各种结构,得到了能量最稳定构型,最稳定结构均也有以下特点为:Cu3团簇中一个Cu原子与苯环平面上1-甲基相邻的β、γ碳原子发生作用新生成Cu-C-C三元环结构,其中邻二甲苯配合物中Cu-C键长分别为 2.227 和 2.216 Å,距甲基较近的键较长,邻二甲苯轨道能级差为 0.1796 a.u., Cu3(1,2-(CH3)2C6H4)的能级差为 0.0110 a.u., 体系的吸附能为58.6 kJ·mol-1. 间二甲苯配合物中Cu-C键长分别为 2.200 和2.222 Å,距甲基较近的键略微短,间二甲苯轨道能级差为 0.171 a.u., Cu3(1,3-(CH3)2C6H4)的能级差为0.0109 a.u., 体系的吸附能为61.1 kJ·mol-1. 对二甲苯配合物中Cu-C键长分别为 2.205 和 2.224 Å,距甲基较近的键略微短,对二甲苯轨道能级差为 0.182 a.u., Cu3(1,4-(CH3)2C6H4)的能级差为 0.0094 a.u., 体系的吸附能为 62.4 kJ·mol-1. 配位反应明显降低了二甲苯体系的能级差, 对进一步催化燃烧将起到显著的促进作用.
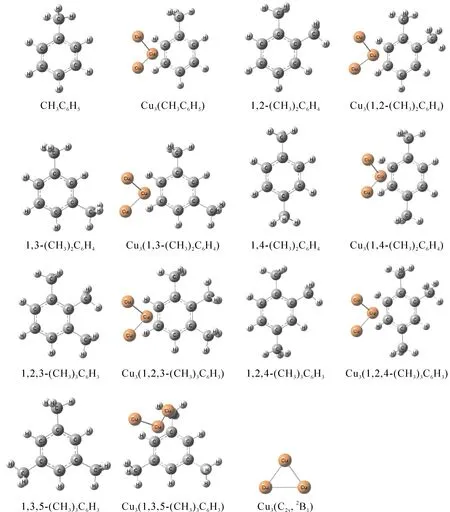
图1 B3LYP/6-311+G(d,p)方法下各分子的基态几何构型 Fig. 1 Geometries of various molecules on the ground state at the B3LYP/6-311+G(d,p) level
3.3 Cu3(CH3)3C6H4配合物
关于Cu3与三甲苯作用体系,我们对系统研究了Cu3与1,2,3-三甲基苯、1,2,4-三甲基苯、1,3,5-三甲基苯三种异构体作用的各种结构,得到了能量最稳定构型,最稳定结构均也有以下特点为:对于1,2,3-三甲基苯、1,2,4-三甲基苯,Cu3团簇中一个Cu原子与苯环平面上1-甲基相邻的 β、γ 碳原子发生作用新生成Cu-C-C三元环结构; 对于1,3,5-三甲基苯,Cu3团簇中一个Cu原子与苯环平面上1-甲基相邻的 α、β 碳原子发生作用新生成Cu-C-C三元环结构, 其中1,2,3-三甲基苯配合物中Cu-C键长分别为 2.216 和 2.225 Å,距甲基较近的键较短, 1,2,3-三甲基苯轨道能级差为 0.1783 a.u., Cu3(1,2,3-(CH3)3C6H3)的能级差为 0.0101 a.u., 体系的吸附能为 61.3 kJ·mol-1. 1,2,4-三甲基苯配合物中Cu-C键长分别为 2.219 和 2.210 Å,距甲基较近的键略微长,1,2,4-三甲基苯轨道能级差为 0.1731 a.u., Cu3(1,2,4-(CH3)3C6H3)的能级差为 0.0103 a.u. 体系的吸附能为 61.3 kJ·mol-1. 1,3,5-三甲基苯配合物中Cu-C键长分别为 2.296 和 2.183 Å, 距甲基较近的键略微长, 1,3,5-三甲基苯轨道能级差为 0.1821 a.u., Cu3(1,3,5-(CH3)3C6H3)的能级差为 0.0094 a.u., 体系的吸附能为 59.4 kJ·mol-1. 配位反应明显降低了三甲苯各体系的能级差, 对进一步催化燃烧将起到显著的促进作用.
4 结 论
采用密度泛函理论的B3LYP方法, 在6-311+G(d,p)基组水平上计算获得了Cu3团簇与甲苯、二甲苯、三甲苯的各异构体分子作用的微观机理, 得到了各体系中结构最稳定构型, 通过结合能、前线轨道等分析苯系物Cu3金属配合物结构与性质, 得到以下结论:各体系中,Cu3团簇中一个Cu原子与苯环平面上相邻两个碳原子发生作用新生成三元环结构,Cu3团簇与甲苯作用的结合能为58.3 kJ·mol-1, Cu3团簇与二甲苯作用过程中,对二甲苯的结合能最高,为62.4 kJ·mol-1,Cu3团簇与三甲苯作用过程中, 1,2,4-(CH3)3C6H3的结合能最高,为62.7 kJ·mol-1. 比较反应物和配合物的前线轨道能级差得到,配位反应明显降低了各体系的能级差, 对进一步催化燃烧将起到显著的促进作用.
猜你喜欢
暨南学报(哲学社会科学版)(2022年3期)2022-11-09
化工管理(2020年11期)2020-04-23
青岛大学学报(工程技术版)(2019年2期)2019-09-10
西部探矿工程(2018年9期)2018-09-11
高校招生(2017年5期)2017-06-30
电子制作(2017年1期)2017-05-17
中国纤检(2017年2期)2017-03-16
中国医学装备(2016年11期)2016-12-09
西南国防医药(2016年6期)2016-12-01
枣庄学院学报(2015年5期)2016-01-09
