
外电场下L-茶氨酸分子的结构特征与光谱性质
2020-04-28 04:19:18于建成唐天宇唐延林
原子与分子物理学报 2020年1期
袁 荔, 施 斌, 于建成, 唐天宇, 袁 园, 唐延林
(贵州大学物理学院, 贵阳 550025)
1 引 言
L-茶氨酸(N-乙基-L-谷氨酰胺)是从茶叶中提取的非蛋白氨基酸,是茶汤滋味品质的重要物质,在茶梅、蘑菇、油茶、红山茶等植物中微量检测到,在其他植物中还未发现[1]. 它具有降血压、抗肿瘤、抗疲劳、抗氧化、静心安神等作用,在医药、食品、保健领域得到了应用[2-7]. 现有文献从茶氨酸及其衍生物的提取、合成、生化作用等角度进行了广泛研究[8-10],但对其理论研究尚未见报道.
电场作用下的分子会产生新的物理现象和化学变化,比如分子结构发生变化,化学键的断裂,激发态的改变,分子极性的变化,光谱红移或蓝移等[11-13]. Yin等人[14]计算处于外场下二噁英的几何构型、能量、偶极矩等参数,发现分子键长发生改变,能量随电场增大而减小,偶极矩随电场增大而增大. 吴学科等人[15]计算在外场中SF6的红外光谱与分子结构,结果发现分子的LUMO能级更易受到外场的影响,能隙随外电场增大而逐渐减小,同时部分无红外活性的模式在外场下具有了活性等. 林华等人[16]计算处于外电场中二甲苯分子结构与光谱的变化特性,其总能量,能带间隙随电场先增加后减小,并关于F=0这个轴对称,紫外-可见光谱随着外电场的增加逐渐红移等.
本文采用密度泛函理论(DFT),在B3LYP/6-311g(d,p)的水平上,计算不同外电场(0~0.0125 a.u.,0~6.428×109V/m)对L-茶氨酸分子的光谱性质和结构特征的影响,分析处于电场中分子的红外光谱变化趋势,电场如何影响分子的结构,紫外光谱在电场中的变化情况等. 为L-茶氨酸的的光谱检测、光谱分析、物质合成及提取应用提供理论依据.
2 理论与计算方法
在外电场作用下,分子体系哈密顿量H具有下列形式:
H=H0+Hint
(1)
H0是无外场时的分子哈密顿量,Hint是外电场与分子相互作用时的哈密顿量[17]. 当考虑体系中电场与分子相互作用,并在偶极近似下,Hint与电场强度F具有以下关系:
Hint=-μ·F
(2)
本文使用Gaussian09软件包对L-茶氨酸分子进行理论计算,采用B3LYP方法,6-311g(d,p)基组对分子几何优化,并在相同的水平上计算处于不同外电场下的分子几何构型、激发态、频率、前线轨道的能量与能隙. 使用Tian和Chen[18]开发的Multiwfn软件分析前线轨道,激发态及光谱. 对优化后的L-茶氨酸分子在x轴方向上加0~0.0125 a.u.的外电场,如图1所示.
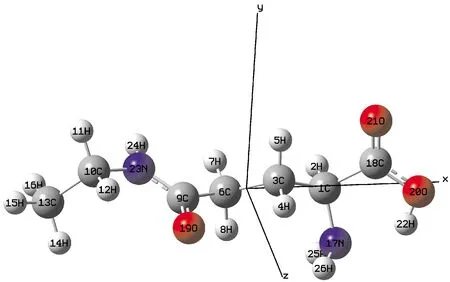
图1 优化的L-茶氨酸分子的几何结构Fig.1 Optimized geometric structure of L-theanine molecule
3 结果与分析
3.1 不同外电场下的L-茶氨酸几何构型
基于DFT/B3LYP方法,在6-311g(d, p)基组上优化处于不同外电场(0~0.0125 a.u.)的L-茶氨酸的几何基态结构. 表1显示出不同外场下的键长变化,由于篇幅关系,表中仅列出主要的键长,随着电场的增大,键长RC-C(6,9),RC-O(9,19),RN-H(17,25),RN-H(17,26),RC-O(18,21),RO-H(20,22)逐渐增大,RC-N(9,23)逐渐减小,RC-N(1,17)先减小后增大. 总的依据是:分子的稳定状态由分子内电场与所加外场共同决定的,原子周围的电子云随着所加的电场产生了逆电场方向偏移现象[19], 形成与外电场方向相同或相反的内电场,导致键长的减小或增大. 无电场时,RC-C(6,9),RC-O(9,19),RC-O(18,21)的键长小于邻近的键长RC-N(9,23),RC-O(18,20),邻近键长更易受到电场的影响,当邻近的键长随电场减小时,RC-C(6,9),RC-O(9,19),RC-O(18,21)键长被动增大.RC-N(1,17)先减小后增大是因为N的电负性比C的强,吸引电子的能力也就比C强,内电场方向由C指向N. 当电场小于0.0025 a.u.时,内电场方向与外电场方向成锐角,电场增强,RC-N(1,17)减小;当电场大于0.0025 a.u.时分子发生弯曲,使得1C-17N的内电场方向与外场方向呈钝角,与外电场方向相反,从而键长增大,所以RC-N(1,17)先减小后增大. 且当电场强度达到0.0125 a.u. 时,分子中羧基上的O-H发生断裂. 表2显示出分子键角、二面角随电场的变化,在外场作用下,分子发生的弯曲与折叠.
表1 不同外加电场下L-茶氨酸的键长
Table 1 The bond lengths of L-theanine under different external electric fields
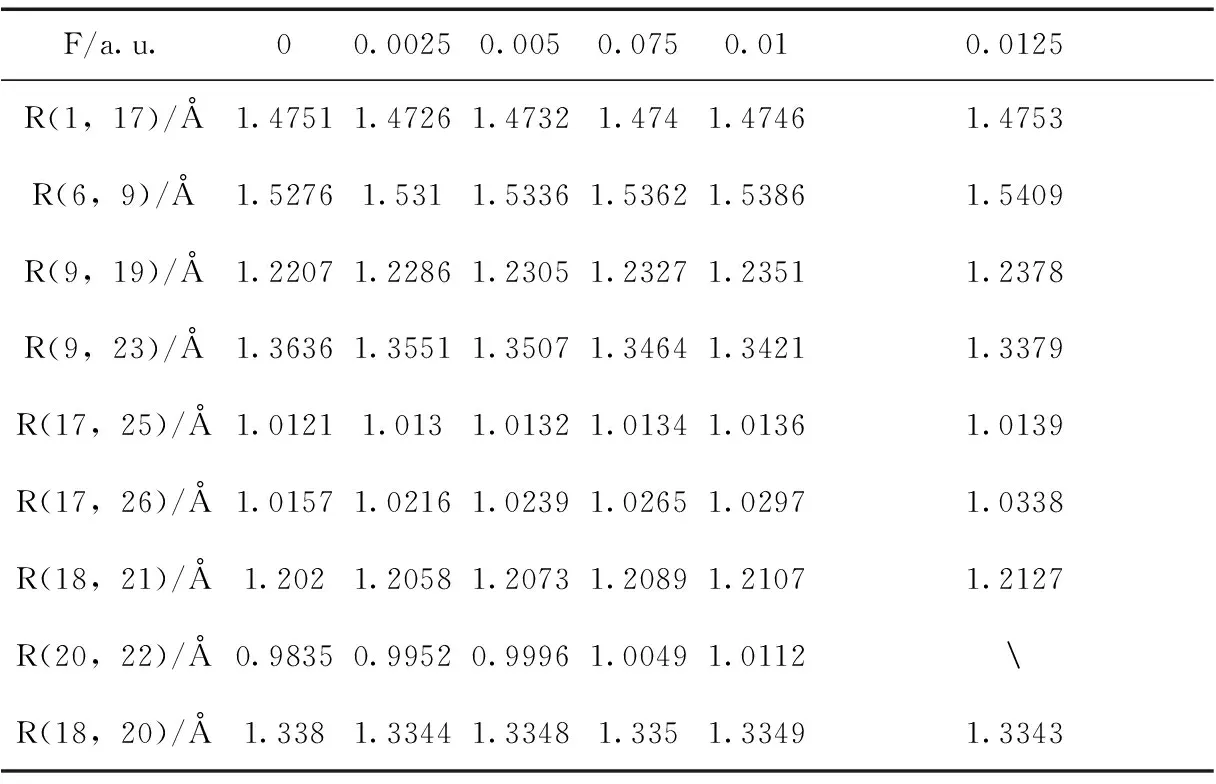
F/a.u.00.00250.0050.0750.010.0125R(1,17)/Å 1.47511.47261.47321.4741.47461.4753R(6,9)/Å1.52761.5311.53361.53621.53861.5409R(9,19)/Å1.22071.22861.23051.23271.23511.2378R(9,23)/Å1.36361.35511.35071.34641.34211.3379R(17,25)/Å1.01211.0131.01321.01341.01361.0139R(17,26)/Å1.01571.02161.02391.02651.02971.0338R(18,21)/Å1.2021.20581.20731.20891.21071.2127R(20,22)/Å0.98350.99520.99961.00491.0112R(18,20)/Å1.3381.33441.33481.3351.33491.3343
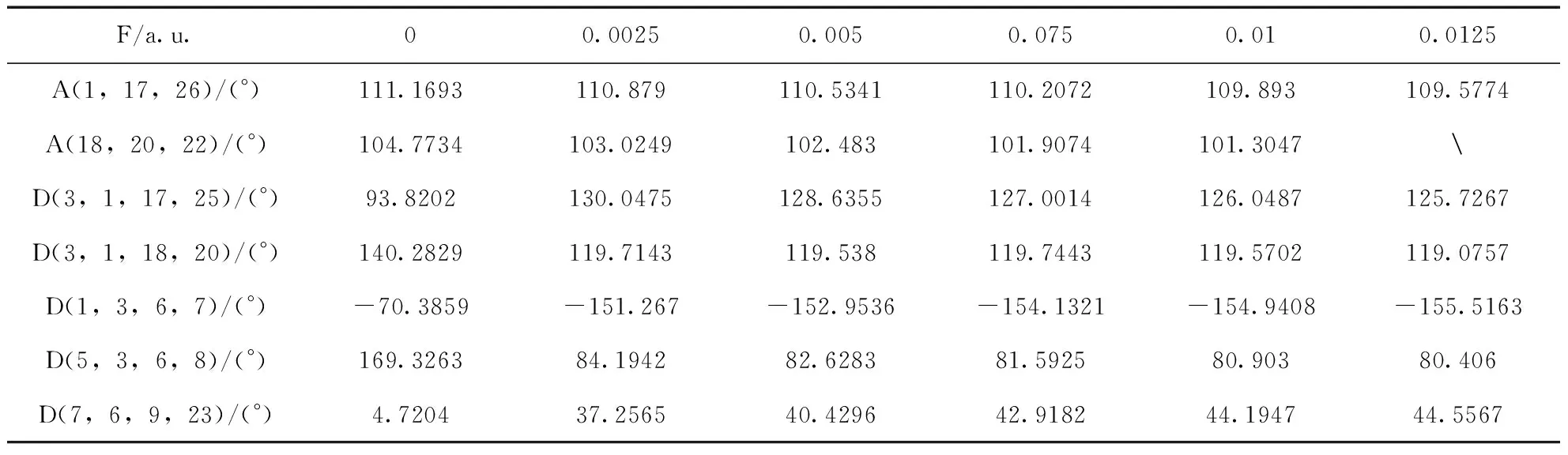
表2 不同外电场下L-茶氨酸分子的键角、二面角
3.2 不同外电场下L-茶氨酸的紫外-可见光谱及前线轨道
基于L-茶氨酸分子的基态几何结构优化,使用TDDFT方法,B3LYP/6-311g(d,p)基组在气相环境下计算了分子的前15个激发态,得到处于不同电场下(0~0.01 a.u.,0.0125 a.u. 时分子发生键的断裂)L-茶氨酸分子的紫外(UV)吸收光谱,如图2所示. 当电场增加时,紫外吸收光谱的摩尔吸收系数、线宽随电场改变,光谱先蓝移再红移. 从图3和表3可知,在加大外场的情况下,最高占据轨道的能量(EHOMO)先增加后减小再增加,最低空轨道的能量(ELUMO)先小幅度减小后逐渐变大,能带间隙(EG)先增大后减小.
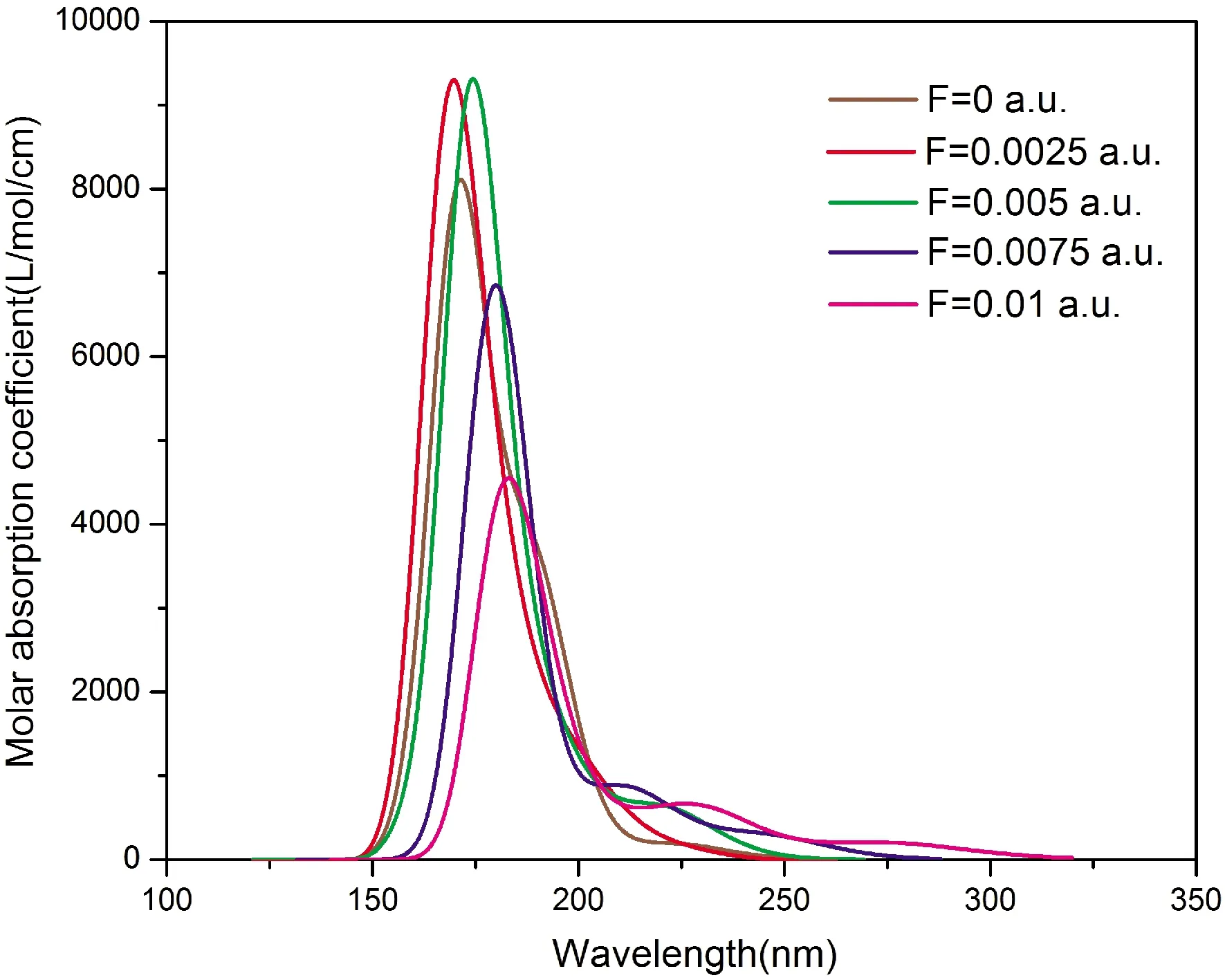
图2 不同外电场下L-茶氨酸分子的紫外吸收光谱Fig. 2 Ultraviolet absorption spectra of L-theanine molecules under different external electric fields
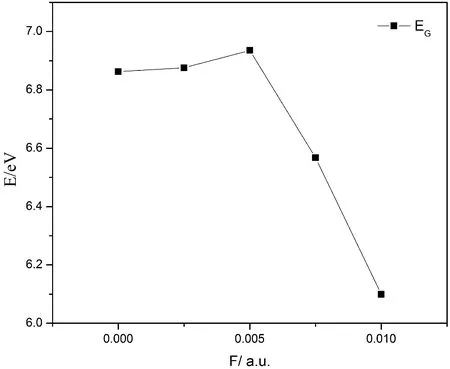
图3 不同外电场下L-茶氨酸分子的能隙Fig.3 Energy gaps of L-theanine molecule under different external electric fields
F=0 a.u.,紫外吸收光谱由基态S0跃迁到激发态S3、S4、S5、S9、S10、S11、S12、S13、S15的贡献得到;F=0.0025 a.u.时,吸收光谱主要是由基态S0跃迁到激发态S3、S4、S5,S7、S9、S10、S11、S12、S13、S14得到;F=0.005 a.u,吸收光谱主要由S0跃迁激发态到S6,S8,S10,S11、S13、S15得到;F=0.0075 a.u.时,吸收光谱主要由基态S0跃迁到激发态S6、S10、S11、S12得到;F=0.01 a.u. 时,吸收光谱主要由基态S0跃迁到激发态S9,S13,S14,S15得到.
表3 不同外电场下L-茶氨酸分子的前线轨道能量
Table 3 The frontier orbital energies of L-theanine molecule under different external electric fields

F/a.u.00.00250.0050.00750.01EHOMO/eV-6.966127-6.85483-7.027997-6.898006-6.697712ELUMO/eV-0.1037210.020788-0.092463-0.330839-0.59847
电场不仅改变激发态的跃迁情况,同时还改变激发态轨道的组成成分,使得在不同的电场中,激发态能量随之不同. 例如,当F=0 a.u.和F=0.005 a.u.时,分子紫外吸收光谱的中心波长都主要由基态跃迁到第 10激发态得到,但在F=0 a.u时,第10激发态中的占比最大的轨道跃迁是从HOMO-3轨道跃迁到LUMO轨道,F=0.005 a.u.时,第10激发态中占比最大的轨道跃迁是从HOMO-1轨道跃迁到LUMO+2轨道,所以激发态能量产生差异,随之激发态与基态之间的能隙不一样,紫外吸收光谱随能隙的变化而变化,从表4可知,中心波长对应的激发态能隙先增加后减小,所以紫外吸收光谱先蓝移再红移.
"content">表4 不同外电场下紫外吸收光谱中心波长对应的跃迁激发态间能隙
Table 4 Energy gaps between transition excited states corresponding to the central wavelength of ultraviolet absorption spectrum under different external electric field

λ/nm173.1171.12174.79179.53183.04△E/eV7.16247.24547.09346.90626.7737
摩尔吸收系数在电场小于0.005 a.u. 时增加,大于0.005 a.u.、小于0.01 a.u. 时变小,谱线宽度先变窄后变宽. 这主要是因为摩尔吸收系数与跃迁概率呈正相关关系,跃迁概率越大,摩尔吸收系数随之增大. 外场改变了分子从基态跃迁到各个激发态的跃迁几率,所以摩尔吸收系数随电场发生变化. 从不确定关系:ΔE·Δt≥ħ可知,光谱线宽(ΔE)与激发态寿命(Δt)呈反比关系,外场影响到分子激发态的寿命,从而紫外吸收光谱线宽随着外场而改变.
从前线轨道理论可知,当分子受到小于0.005 a.u.的电场时,前线轨道能带间隙增大,电子跃迁的能力减弱,分子更为稳定,但当分子受到更大的外电场时,能带间隙急剧减小,分子易从HOMO轨道受激跃迁到LUMO轨道,分子活性增强,可通过调整外加电场强度去改变分子的稳定状态.
3.3 不同外电场下L-茶氨酸的红外光谱
在B3LYP/6-311g(d, p)几何优化的基础上进一步优化计算了处于不同外电场(0~0.01 a.u.)下L-茶氨酸分子的频率,如图4-a所示. 外加电场等于0时,光谱图上频率为3349 cm-1主要对应着羟基上O-H的伸缩振动,1788 cm-1主要对应羧基上18C=21O的伸缩振动及其引起的整个羧基的振动,1696 cm-1主要由酰胺上9C=19O的伸缩振动及其引起的周围部分原子的振动产生的频率,1496 cm-1主要对应酰胺基团中23N-24H的面内摇摆,1384 cm-1主要是20O-22H的弯曲振动,810 cm-1对应着氨基17N-25H-26H的摇摆振动,446 cm-1是酰胺中23N-24H的面外摇摆振动.
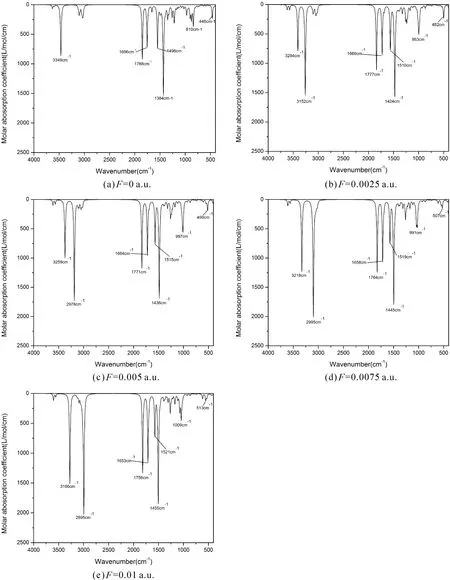
图4 不同外电场下L-茶氨酸的红外光谱图Fig.4 Infrared spectrograms of L-theanine under different external electric fields
当所加电场逐渐增大时,红外光谱3294 cm-1波数对应的氨基17N-25H-26H的伸缩振动振幅明显增大,同时在分子对应的主要伸缩振动模式上(3349 cm-1、3294 cm-1、1788 cm-1、1696 cm-1)发生显著的红移,在分子主要弯曲振动模式上(1496 cm-1、1384 cm-1、810 cm-1、446 cm-1)发生轻微的蓝移. 分子的红外振动频率以及振动幅度易受到外电场影响. 外电场下9C=19O,17N-25H,17N-26H,18C=21O,20O-22H键长增长,所以对应的振动吸收峰红移;光谱蓝移处于分子(23N-24H、20O-22H、17N-25H-26H)弯曲振动区域,该区域光谱复杂,易受到骨架振动和环境变化的影响,外加电场改变了分子的键角和二面角,分子发生弯曲,所以频率蓝移.
4 结 论
本文采用B3LYP/6-311g(d, p)方法优化计算了不同外电场下(0~0.0125 a.u.)L-茶氨酸分子的几何参数,频率,激发态及前线轨道,讨论了外电场对分子键长,红外、紫外吸收光谱,前线轨道能量及能隙的影响.
随外电场增强,原子周围的电子云朝逆电场方向重新分布,内电场方向随电子云的变化而变化,从而9C-23N键长减小;17N-25H、17N-26H、20O-22H键长增长;6C-9C、9C=19O、18C=21O键长受到邻近键长的影响而被动增长;1C-17N键长先减小后增大,原因是分子骨架在外场的影响下发生弯曲,导致1C-17N的内电场方向发生了变化. 当外电场为0.0125 a.u.时,20O-22H键断裂,分子解离. 在红外光谱中,分子的伸缩振动模式(3349 cm-1、3294 cm-1、1788 cm-1、1696 cm-1)发生红移,原因是模式对应的9C=19O,17N-25H,17N-26H,18C=21O,20O-22H键长随外电场增大而增长;分子的弯曲振动模式(1496 cm-1、1384 cm-1、810 cm-1、446 cm-1)发生蓝移,原因是分子骨架在电场下产生形变. 电场影响分子激发态跃迁,随外电场的增加,其紫外光谱中心波长对应的跃迁激发态能隙先增加再减小,所以紫外吸收光谱先小幅蓝移后再逐渐红移,同时外电场影响分子的跃迁概率和激发态寿命,使得紫外吸收光谱的幅度和谱宽随之变化.
猜你喜欢
吉林大学学报(理学版)(2025年1期)2025-02-06 00:00:00
中国交通信息化(2021年1期)2021-06-11 01:23:46
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
中国交通信息化(2017年1期)2017-06-08 06:05:04
中国交通信息化(2017年4期)2017-06-06 07:21:48
中国交通信息化(2017年10期)2017-01-14 17:06:07
陕西理工大学学报(自然科学版)(2015年4期)2016-01-16 03:05:41
中学化学(2015年8期)2015-12-29 07:32:44
原子与分子物理学报(2015年3期)2015-11-24 12:49:36
原子与分子物理学报(2014年1期)2014-03-20 08:16:14
