
热效应对tP10-FeB4的电子结构与光学性质的影响
2020-04-28 04:20董明慧苑光明尹田田张彩丽
原子与分子物理学报 2020年1期
董明慧, 苑光明, 尹田田, 张彩丽
(1. 齐鲁理工学院, 济南 250200; 2. 太原理工大学材料科学与工程学院, 太原 030024)
1 Introduction
Metal borides for their desirable characteristics such as higher strength, outstanding hardness, good thermal and chemical stability[1], have extensive applications, such as Nd2Fe14B which is the best permanent magnet, MgB2which is a the first metallic superconductor, and YB66used as monochromator for soft synchrotron radiation[2]. For this phenomenon there are two main reasons. On the one hand, metal borides, especially transition-metals (TM) borides, usually have high valence electron densities, which are easy to form strong covalent bonds. On the other hand, the discovery of superhardness metal borides such as FeB4has stimulated people to search for new superhard materials in transition-metals borides. Therefore, a large number of theoretical and experimental studies on metal borides have emerged. Up to now, Cr, Mo, W, Tc, Ru, Os, Nb, Ti, Ir, Mn, Fe, etc were found to have the ultra-incompressible and expected to have good mechanical properties[3-10].
Among these advanced matetials, FeB4, because of the outstanding hardness and semiconducting characteristics, attracted much more attention. Kolmogorovetal.[11]reported two stable phases of FeB4, i.e.,oP10-FeB4andoP12-FeB2. Bialonetal.[12]demonstrated thatoP10-FeB4is stable under high pressure. However, Wang[13]reported thatoP10-FeB4is only a hard material but not a superhard material.Wang[14]proposed the alternative structuretP10-FeB4is a superhard material which hardness reaches 45.4 Gpa. FeB4is often employed in industry as protective coatings on steels to increase wear resistance which means high pressure and temperature[14]. Unfortunately, Until now, some of their properties are under ideal conditions[14,15]. Therefore, it is necessary to study the effect of temperature on the properties of FeB4.
2 Computational methods
All of the calculations were performed using CASTEP[16, 17]based on DFT[18]. Generalized gradient approximation (GGA) and the Perdew-Burke-Ernzerhof (PBE)[19]function were employed as exchange-correlation potential. The interaction between valence electron and ion core was ultrasoft pseudopotential. The valence electron configurations for Fe and B were considered as 3p63d74s1and 2s22p1, respectively. The cut off kinetic energy was set as 380 eV and Monkhorst-Pack[20]k-points were chosen to 6×6×6 in the Brillouin zone to ensure that all of the calculations were converged. The convergence of the total energy was taken as 1.0×10-5eV/atom, the interaction force between atoms was less than 0.03 eV/atom, the maximum displacement was 0.001 Å. The spin polarization effect was considered due to the partial occupations of 3d-orbitals of Fe.
3 Results and discussion
3.1 Structural properties
The tP10-FeB4crystal has tetragonal structure in space groupP42/nmc. As shown in Fig. 1, each unit crystal contains two Fe atoms and eight B atoms. In the crystal, B (8g) atoms occupy (0.250, 0.5, 0.628) position and Fe (2b) atoms occupy (0, 0, 0.5) position.

Fig. 1 The crystal structure oftP10-FeB4
The crystal structure was fully optimized with respect to determine the lattice parametersa,b, andc. As shown in Table 1, the calculated lattice constants match well with the available previous theoretical values. The deviations between our theoretical values and Wang[22], Zhao[15], Huang[20]are all less than 1%. However, the lattice parameters researched by Kotmool[20]are about 5%. This is because Kotmool predicted the structure by the evolutionary algorithm (USPEX)[23,24]. By comparison, we find that DFT is more reliability than USPEX. Therefore, in order to ensure the correctness and reliability of the results, DFT method was used in our calculation.
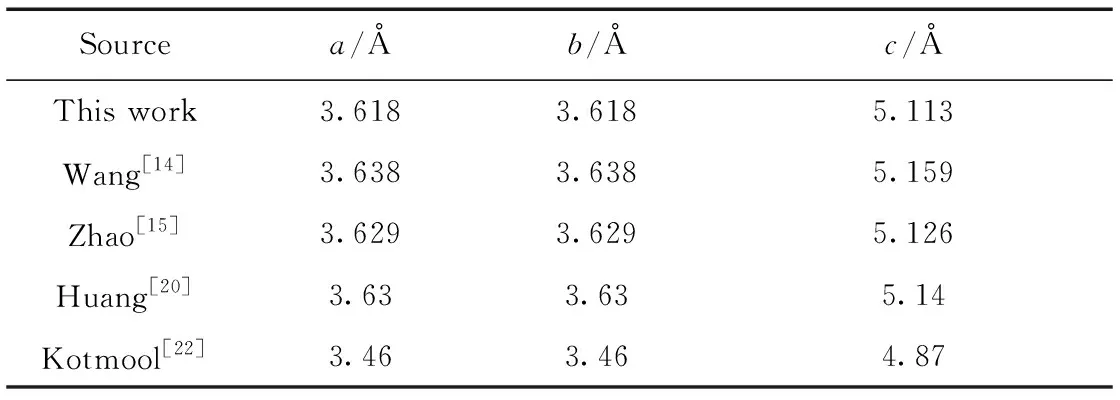
Table 1 Calculated equilibrium lattice parameters a, b, c
In addition, the band gap width (Eg) oftP10-FeB4 in ideal state was also studied. The calculated result (Eg=1.773 eV) is in good agreement with that in the literature (1.85 eV[14]). However, it is slightly lower than GW algorithm (2.43 eV[15]). This is mainly due to the energy gap width obtained by using density functional theory[20].
3.2 The influence of temperature on electronic properties of tP10-FeB4
In order to study the effect oftemperature on the electronic structure oftP10-FeB4, the calculation changes from simple structural optimization to molecular dynamics simulation. The model used in the calculation is the cell optimized before. The NPH ensemble is used in the calculation process. BecausetP10-FeB4is mainly used in 250-350 K temperature range, therefore, the temperature of calculation process is set as 250 K, 280 K, 310 K and 340 K temperature, respectively. Other parameters such as integration step time, total time, and iteration step are set as 1 fs, 0.5 ps, and 1000 steps, respectively.
Fig. 2 shows that the increase in temperature could shorten band gap width (Eg) slightly (less than 6%). This means that the electronic structure oftP10-FeB4could withstand higher temperature which is attributed to the strong bonding between Fe and B atoms.

Fig. 2 The calculated band gap width at different temperatures
Fig. 3 is the density of states (DOSs) of Fe atoms at 250 K, 280 K, 310 K and 340 K, respectively. It can be seen from the figure that the valence band (VB) shifts towards higher energy level with the increase of temperature, which is mainly due to the higher energy level occupied by electrons. The valence band maximum (VBM) and conduction band minimum (CBM) of Fe are mainly composed of Fe-3d. Corresponding to different temperatures (250 K, 280 K, 310 K and 340 K), the Fe-3delectronic states near the VBM mainly distribute between -10.25~0 eV, -9.86~0 eV, -8.86~0 eV and -7.52~0 eV, respectively. In the meanwhile, the Fe-3pelectronic states near VBM mainly distribute between -3.32~0 eV, -2.51~0 eV, -2.32~0 eV, -2.11~0 eV, respectively. The peak width of Fe-3delectronic states near the VBM becomes narrow with the increase of temperature. Therefore, the dispersion of the electronic states becomes weakened and localization enhanced. Simultaneously, the conduction band (CB) also tends to shift towards the higher energy level with the increase of temperature. However the influence of temperature on CB is not obvious. Therefore, the work together of VB and CB results in band gap width narrowing.

Fig. 3 DOSs of Fe at different temperatures
Fig. 4 shows the DOSs of B atoms at 250 K, 280 K, 310 K and 340 K, respectively. For the B atom, the VB shifts to higher energy level with the increase of temperature. The VBM and CBM of B are mainly composed of B-2pelectronic states. Corresponding to different temperatures (250 K, 280 K, 310 K and 340 K), the B-2pelectronic states near the VBM mainly distribute between-12.97~0 eV, -11.58~0 eV, -10.35~0 eV, and -10.05~0 eV, respectively. The distribution of B-2selectronic states are the same as B-2p. Such as the atom of Fe, the CB also tends to shift towards the higher energy level with the increase of temperature. However the influence of temperature on CB is not obvious. In addition, we find that the B-2pelectronic states are very similar to the Fe-3delectronic states in the vicinity of Fermi energy by comparing Fig.3 and Fig.4. Therefore, it can be conclude that strong orbital hybridization exists between atom Fe and B.

Fig. 4 DOSs of B at different temperatures
3.3 Thermal effect on the optical properties of tP10-FeB4
As we all know, the interaction of a photon with the electrons in the semiconductor system can lead to the transitions between occupied and unoccupied states. In the linear response range, the optical material properties can be determined from the dielectric function[25-27]
ε(ω)=ε1(ω)+iε2(ω)
(1)
(2)
δ[EC(k)-EV(k)-ħω]
(3)
(4)
(5)
In theses equations,VandCrepresent valence bands and conduction bands, respectively.BZrepresents the first Brillouin zone,ωis angular frequency,kis the inverted sagittal, andEv(k) ,Ec(k) are the energy levels which located at valence band and conduction band, respectively. From the above functions(1)-(5), the optical properties, such as the absorption coefficient, reflectivity index are derived from the dielectric function. Equations (2) and (3) are also called Kramer-Kronig transformation. It must be mentioned that all of the calculations in this paper were based on the DFT. However, experience has proved that the energy gap width calculated by DFT is smaller than that obtained by experiment[20]. In order to overcome this defect, scissors operator is usually used to correct the results in optical property analysis[28, 29]. In this paper, the energy gap width is 1.773 eV, while the reported is 2.43 eV[15]. Therefore, 0.567 eV was chosen as scissors operator.
The calculated imaginary partε2(ω) and real partε1(ω)of dielectric function with a range of 0~20 eV fortP10-FeB4are shown in Fig. 5. It is worth noting that with the increase of temperature, main two peaks could be observed in imaginary partEAand real partEC: 5.32 eV and 3.57, 6.12 eV and 4.42 eV, 6.83 eV and 5.16 eV, 7.48 eV and 5.83 eV. In consideration of the DOSs, the peak A and C relate to the transition from the Fe-3din VBM to the B-2pin CBM. In addition, with the increase of temperature, the peak value increases slightly. This is consistent with the Fe-3dpeak value changing regulation in the VBM. Fig. 4 real part is theε1(0) of dielectric function fortP10-FeB4crystals. The calculated static dielectric constantsε1(0) for 250 K, 280 K, 310 K, 340 K are 13.24, 14.53, 15.02 and 16.32, respectively. It can be seen from the real part of Fig.5 that the values ofε1(0) increase gradually with the increase of temperature. However, it reveals that the obtainedε1(0) has inverse relation to the calculatedEg, which agree well with the relationship[30]of equation (6).
(6)
According to Lambert-Beer’s law[31], the absorption coefficientI(ω)is proportional to the absorbanceA(ω). The absorption coefficientI(ω)oftP10-FeB4is plotted in Fig. 6. At the beginning, the absorption remains at a low level. Then the absorption gradually increases after 2.5 eV, reaching 28500 cm-1at 6.33 eV, above the end of the ultraviolet region (3.1 eV-6.2 eV). Therefore, the absorption coefficient in ultraviolet region is excellent. After the first peak, the change of absorption coefficients become weak, and the maximum value of absorption coefficient appears at about 15.5 eV. In addition, it can be found that with the increase of temperature, the redshift phenomenon appears. This is also consistent with the band gap width change regulation. All of the above imply thattP10-FeB4could be used as selective ultraviolet absorption materials.


Fig. 5 The dielectric function oftP10-FeB4
From the Fig.7, we know that the reflectivity index at 0 eV is 0.37, and then increases to the first peak appears at about 4.95 eV, however, the change is slightly. What’s more, it can be found that the redshift phenomenon alsoappears with the increase of temperature. However, in the ultraviolet region, the effect of temperature on reflectivity index is also not obvious.

Fig. 7 The reflectivity index oftP10-FeB4
4 Conclusions
The thermal effects on the electronic and optical properties of thetP10-FeB4were researched using first-principles based on DFT. The followings are the key findings.
1.The increase in temperature could shorten band gap width oftP10-FeB4slightly, which means that the electronic structure oftP10-FeB4could withstand higher temperature.
2.The analysis results of DOSs show that a strong covalent bond is formed between the Fe and B atoms, contributed to improve the stability of the structure. According to the DOSs, the VB and CB are dominated by Fe-3dand B-2pstates. The band gap width decreases with temperature increasing .
3.With the increase of temperature, the redshift phenomenonappears in absorption spectrum and reflectivity spectrum, which is consistent with the band gap width change regulation.
4. In the ultraviolet region,tp10-FeB4has a good absorption spectrum and is a kind of potential ultraviolet absorption material.
猜你喜欢
天津建设科技(2022年3期)2022-06-22
大学生(2021年9期)2021-09-28
科技进步与对策(2020年12期)2020-07-03
——材料科学与工程
湖南人文科技学院学报(2020年3期)2020-06-08
水利规划与设计(2020年1期)2020-05-25
中学生数理化(高中版.高二数学)(2020年2期)2020-04-21
校园英语·上旬(2019年6期)2019-10-09
山东开放大学学报(2019年3期)2019-08-02
成都信息工程大学学报(2019年1期)2019-05-20
中国机械工程(2018年21期)2018-11-13
