
TUBA1A 基因突变致大脑皮层发育不良2 例临床及遗传学分析
2020-04-27 05:37:56黎珍妹赵红玲余红蕾
临床儿科杂志 2020年4期
张 潮 黎珍妹 赵红玲 李 雯 余红蕾
咸宁市中心医院 湖北科技学院附属第一医院儿科(湖北咸宁 437100)
人类大脑皮层的发育是一个复杂而精密的过程,发育过程中任何一环节出现问题均可以引起发育落后、智力低下、癫痫发作等一系列的临床表现[1]。而神经元移行相关基因突变是大脑皮层发育不良(malformation of cortical development,MCD)的重要原因[2]。研究发现,Reelin-LIS1-Tubulins信号通路可调控神经元移行,该通路上的多个基因异常可导致不同类型的MCD,其中TUBA1A基因编码微管相关蛋白,该基因突变可导致平滑脑畸形、小头畸形以及多小脑回畸形等[3-4]。TUBA1A基因变异导致的MCD国内未见报道,现报告2例TUBA1A基因变异导致的MCD患儿的临床资料,并复习相关文献,探讨其临床特点。
1 临床资料
例1,男,1岁3个月,确诊癫痫1个月,3天内再次发作10余次就诊。患儿1个月前无明显诱因突发抽搐,强直阵挛发作,具体表现为双眼凝视、口唇面色发绀,口吐白沫,四肢强直,意识丧失,约1~2分钟自行缓解,缓解后精神状态差。考虑癫痫给予左乙拉西坦,早晚各1.5 mL(约37.5mg/kg·d),用药后无抽搐发作。3天前患儿再次出现抽搐,3~5次/d,发作表现与之前类似,门诊以癫痫收入院。患儿为G2P2,足月剖宫产,出生体质量3.1 kg。患儿整体发育落后,独站不稳,精细动作明显落后同龄儿童。入院体格检查:面容无异常,神志清楚,颈软,呼吸运动对称,心脏各听诊区未闻及杂音,肌张力偏低,布氏征阴性。实验室检查:血常规、肝肾功能均未见异常,血氨基酸代谢物未见明显异常。脑电图示有癫痫波发放。头颅磁共振成像(MRI)示神经元移行障碍(平滑脑、巨脑回)。见图1。患儿出院后随访暂未出现癫痫发作。
例2,女,6月龄,因间断抽搐3天入院。患儿无明显诱因出现抽搐,表现为突然点头、上肢内收,成串发作;四肢无明显抖动及强直,口唇面色无明显异常,症状持续数秒至2分钟左右,无发热、无吐泻。患儿为G2P1,足月剖宫产,出生体质量3.4 kg,发育无明显异常,无特殊疾病史,无类似疾病家族史。入院体格检查:面容无异常、神志清楚、全身皮肤未见皮疹;双肺呼吸音稍粗,未闻及干湿性啰音;心率齐,未闻及病理性杂音;腹部无异常;四肢未见异常且活动不受限,肌张力无异常,四肢肌力四级,布氏征及巴氏征阴性。脑电图示异常幼儿脑电图,高度失律并检测到孤立或成串痉挛发作。MRI示双侧额顶叶巨脑回畸形(图1)。患儿入院后予口服妥泰及开普兰治疗,未见好转;随后加抗癫痫药物德巴金并予以静脉滴注ACTH一周(因肺炎停用),之后2 个月偶有发作。目前患儿使用泼尼松龙治疗,抗癫痫药物为丙戊酸钠早晚4 mL,约30mg/(kg·d);托吡酯早晚各12.5 mg,定期随访中。
为进一步明确诊断及寻找病因,经医院医学伦理学委员会审核及家属知情同意后,分别采集患儿及家属的外周静脉血各2 mL,利用全血基因组DNA提取试剂盒(天根生物)提取基因组DNA,委托第三方公司进行全外显子测序,并利用Sanger测序验证。致病基因TUBA1A的引物由上海生工合成。经过高通量测序以及Sanger 测序验证后发现,例1 的TUBA1A基因存在c.387 C>A 杂合突变,该突变导致该基因编码的蛋白第129 位氨基酸由半胱氨酸突变为终止密码子(p.C 129 X);患儿父母该位点未见异常,见图2。例2的TUBA1A基因存在c.848 A>G 杂合突变,该突变导致该基因编码的蛋白第283位氨基酸由组氨酸突变为精氨酸(p.H283R),患儿父母该位点未见异常,见图2。

图1 患儿头颅MRI 表现

图2 2 例患儿TUBA1A 基因测序图
2例患儿TUBA1A基因存在的2种突变,c.387C>A和c.848 A>G 均未见报道。其中c.387 C>A 为无义突变,依据ACMG指南为致病性改变(PVS1,LOF变异可能导致基因功能丧失;PS2:新发突变,无家族史;PM1:数据库未收录,属于低频变异)。c.848A>G为错义突变,Polyphen2及SIFT软件预测突变影响蛋白质的功能不同物种间蛋白序列保守性分析发现该位点十分保守,见图3。依据ACMG 指南该突变为可能致病突变(PS2:双亲验证的新发突变;PM2:EXaC及其他人群数据库未见收录;PP3多种软件预测有害)。
取患者新鲜静脉血1 mL,裂解红细胞后得到白细胞,利用Trizol 方法提取总RNA,经过反转录得到cDNA,利用TUBA1A基因特异性生物引物进行相对定量分析,结果显示相对于正常对照,例1的TUBA1A基因表达水平显著下降,例2表达水平未见明显异常,见图4。

图3 突变位点在蛋白结构中的位置以及c.848A>G(p.H283R)保守性分析
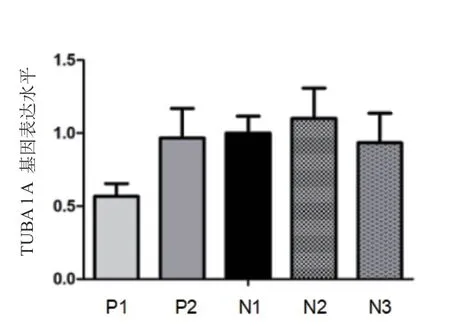
图4 患儿及同龄健康儿童静脉血中TUBA1A 基因mRNA 水平
2 讨论
人体中枢神经系统正常发挥功能需要脑发育早期的两个重要的事件:神经元干细胞的生长分化以及特定的神经元在特定的时间内移行到目的区域并彼此交联发挥正常功能[5]。脑皮层的多层结构就是神经元正常移行形成的,否则会导致脑皮质发育畸形[6]。神经元移行相关基因变异是导致大脑皮层发育不良的重要原因。研究发现,LIS1基因以及DCX基因异常可导致经典型的平滑脑畸形,ARX基因突变可导致X连锁平滑脑畸形伴生殖器异常;FLNA以及ARGEF2基因缺陷可导致脑室周围灰质异位以及可调控神经元移行的Reelin-LIS1-Tubulins信号通路上的多种基因突变可导致MCD[1,7-9]。
本组2 例患儿均以间断抽搐入院治疗,面容均未见异常,入院后生化检测未见明显异常,血氨基酸代谢未见异常。2 例患儿的脑电图均存在异常,高度失率或者癫痫波发放。例1 头颅MRI 提示平滑脑、巨脑回畸形,例2 头颅MRI 示双侧额顶叶巨脑回畸形;均存在MCD的症状。全外显子测序分析发现,2例患儿TUBA1A基因均存在变异,例1为无义突变(p.C129X),导致编码的蛋白质提前终止而不能行使正常功能;例2为错义突变(p.H283R),因TUBA1A蛋白在不同物种间十分保守,推测变异影响蛋白的正常功能。
TUBA1A基因位于12 号染色体q 13.12 区域,全长4530 bp,编码含有451 个氨基酸的α 微管蛋白,该蛋白分为三个结构域,分别为N 端结构域、中间以及C 端结构域[10]。所有真核生物中都有α、β 和γ微管蛋白家族的基因,α 和β 微管蛋白是微管的主要成分。近年发现,越来越多的微管相关的基因(如TUBA1A、TUBA8、TUBB2A、TUBB3、TUBB、TUBB2B等)可导致微管疾病,该类疾病临床表现为巨脑回畸形、多小脑回畸形、胼胝体发育不良、小头畸形、智力低下等一系列症状[11-13]。
文献报道,超过95%的TUBA1A基因突变患者有大脑皮层发育异常的特征,其中约70% 患者为无脑回/巨脑回畸形,约18%存在多小脑回畸形症状。进一步分析发现TUBA1A基因异常可能与基底节、胼胝体以及小脑蚓部异常相关[14]。TUBA1A基因异常导致患者的临床表型多样:超过90%的患者存在全面发育迟缓,约70%存在小头畸形、70%存在不同类型的癫痫发作,约40%肌张力减退,此外少数存在面容异常、斜视、眼球震颤以及视神经发育不全等[14]。本组2例患儿存在MCD、癫痫以及发育落后等与国外报道一致。
进一步检索文献及数据库发现,目前共计收录TUBA1A基因121 个突变,绝大多数为错义突变,其中第420(R402)位以及264(R264)位氨基酸为热点突变位置,20%以上的患者可检测到以上两个热点突变[14-15]。在TUBA1A蛋白的三维结构模型中发现,约40%的致病性突变影响蛋白与其他微管相关蛋白之间或者动力蛋白之间的结合作用。本组例1患儿存在无义突变(p.C 129 X),导致氨基酸编码提前终止,定量PCR 显示TUBA1A的RNA 水平明显降低,推测变异影响蛋白表达量,进而影响功能。例2 携带错义突变(p.H283R),虽变异未影响mRNA水平,推测变异的蛋白与其他微管蛋白的结合作用变弱,进而导致临床表型。遗憾的是,本组2例患儿未检测TUBA1A蛋白的表达水平;未在细胞水平进一步证实突变对TUBA1A蛋白功能的影响。
综上所述,TUBA1A基因突变导致一系列临床表型,包括大脑结构发育异常、整体发育落后,癫痫发作、肌张力异常以及面容、视力异常等,临床诊断较为困难。本组2 例TUBA1A基因异常导致MCD 患儿经分子诊断确诊,且这2 个变异尚未见报道,丰富了TUBAIA基因突变数据库。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
黑龙江大学自然科学学报(2022年4期)2022-11-17 08:07:40
——水芹主要害虫识别与为害症状
长江蔬菜(2022年13期)2022-07-29 01:21:32
中国民间疗法(2021年5期)2021-06-09 09:21:04
中国生殖健康(2020年2期)2021-01-18 02:51:26
小学生导刊(2018年13期)2018-06-29 03:49:00
饮食科学(2017年5期)2017-05-20 17:11:53
中外医疗(2016年15期)2016-12-01 04:25:49
化工进展(2015年6期)2015-11-13 00:29:04
西南军医(2015年4期)2015-01-23 01:19:30
