
超支化聚三唑固态聚电解质及其锂离子和锌离子导电性*
2020-04-19 08:39:28欧笑颖伍建华彭晓春
吉首大学学报(自然科学版) 2020年6期
欧笑颖,刘 奔,伍建华,彭晓春
(1.吉首大学化学化工学院,湖南 吉首 416000;2.吉首大学物理与机电工程学院材料系,湖南 吉首 416000)
具有刚性结构的超支化聚合物具有优良的溶解性,黏度低与力学性能优异,在各类材料领域均有研究[1-2].Cu(I)催化的叠氮-炔点击反应反应条件温和,官能团耐受性好,产率高,在合成超支化聚合物方面有大量应用[3-10].传统基于液态或凝胶态电解质的电池存在漏液问题,安全性低,因此开发安全性较高的固态电解质已成为研究热点[11-12].文献[7]报导了含不同端基的超支化聚三氮唑离子电解质材料,电化学窗口可达到6.0 V(Vs Ag+/Ag),但电导率有待提高(7.7×10-6S/cm).Li[8]在超支化聚三氮唑离子的基础上,引入季铵盐离子,形成杂化聚电解质体系,电导率得到提升(10-5S/cm数量级),电化学窗口分别达到6.0 V(Vs Ag+/Ag)和 4.3 V(Vs Li+/Li).然而这两类超支化聚三唑仍然为粘稠态,不具备可加工性.因此,笔者首先合成两臂含叠氮基单体M1和三臂含炔基单体M2,通过点击化学反应,制备含刚性结构单元的中性超支化聚三唑hb-GPTA,对聚合物的相关性能进行探究,并对聚合物与盐(LiTFSI和Zn(OTf)2)共混体系的电化学性质进行了研究.
1 实验部分
1.1 主要原料
三乙二醇、降冰片烯二羧酸酐、丁二酸酐、抗坏血酸、CuSO4·5H2O、均苯四甲酸二酐、6-氯-1-已醇、4-二甲胺基吡啶(DMAP)、1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐(EDCI)等试剂购买于上海阿达玛斯试剂公司;溴丙炔购买于上海邦成化工有限公司;6-氨基-1-己醇、双三氟甲烷磺酰亚胺锂(LiTFSI)和三氟甲基磺酸锌(Zn(OTf)2)购买于武汉长成化成有限公司;其他试剂均为分析纯.
1.2 结构与性能表征
核磁共振谱(NMR):采用Bruker核磁共振谱仪检测,溶剂为氘代氯仿或氘代二甲亚砜.红外光谱(FTIR):采用Nicolet IS10 FT-IR型傅立叶变换红外光谱仪检测.热失重分析(TGA):采用SDTA851e/SF/1 100 ℃热失重分析仪检测,升温速率为10 ℃/min,氮气氛围,温度范围为30~800 ℃.
电导率的测试:将聚合物与盐的共混物用合适的溶剂溶解,按一定比例混合,待溶剂挥发后,置于真空干燥箱.将带孔(面积为25 mm2)的聚四氟乙烯膜(厚度为90 μm)置于铜片上,聚合物样品滴涂于孔中,置于真空干燥箱,60 ℃干燥4 d,以去除残留水份和溶剂,然后将另一铜片压置于其上,于60 ℃保存1 d,并缓慢降至室温,测试其在30 ℃时的电导率.电导率σ通过以下公式计算:σ=D/(SR),其中,D代表聚合物膜的厚度,S为聚合物膜的面积,R为测得的聚合物本体电阻(通过CHI660C电化学工作站进行交流阻抗谱测得,测试电池采用铜片/聚合物电解质/铜片,振幅:10 mV,频率范围:10 Hz~100 kHz).
电化学窗口的测定:将聚合物制备成标准电池后,利用线性扫描伏安技术(LSV),采用CHI660E型电化学工作站测试.参比电极为不锈钢电极,对电极为锂片或锌片.
2 合成过程
2.1 单体及其中间体的合成
化合物1的合成:
于250 mL三口烧瓶中加入1,6-氯己醇(13.6 g,100 mmol),叠氮化钠(13 g,200 mmol)和DMF(50 mL),90 ℃反应48 h.待冷却至室温后,反应液中加入二氯甲烷,去离子水洗涤,无水硫酸镁干燥,过滤,浓缩,得浅黄色液体12.9 g,收率90.1%.1H NMR(500 MHz,CDCl3),δ: 3.67~3.65(2H,—CH2OH),3.3~3.27(2H,—CH2N3),1.64~1.41(8H,—CH2—).
化合物2的合成:
氮气保护下,于化合物1(4.29 g,30 mmol)的二氯甲烷溶液(30 mL)中加入丁二酸酐(4.5 g,45 mmol)和三乙胺(0.2 g,2 mmol),室温搅拌反应24 h.所得反应液依次用1 M HCl、饱和食盐水洗涤,无水硫酸镁干燥.减压脱除溶剂后,柱层析分离(洗脱剂:V(乙酸乙酯)∶V(石油醚)=4∶1),得到无色黏液6.3 g,收率87.5%.1H NMR(500 MHz,CDCl3),δ:9.22(1H,—COOH),4.13~4.10(2H,—CH2OOC—),3.29~3.27(2H,—CH2N3),2.71~2.62(4H,—CH2CH2COOH),1.67~1.41(8H,—(CH2)4CH2N3).
化合物3的合成[13]:
氮气保护下,于均苯四甲酸二酐(2.5 g,11.5 mmol)的无水DMF(25 mL)溶液中加入6-氨基-1-己醇(3.0 g,25.3 mmol),100 ℃下搅拌反应10 h.反应溶液趁热倒入至70 mL水中,白色固体析出,抽滤,用甲醇洗涤固体,真空干燥,得到白色固体4.78 g,收率98.2%.
单体M1的合成:
冰浴下,于化合物3(1.25 g,3 mmol)的DMF溶液(50 mL)中加入EDCl(1.38 g,7.2 mmol)和DMAP(0.15 g,1.2 mmol),搅拌15 min.将化合物2(2.19 g,9 mmol)的DMF溶液(10 mL)滴入上述体系,自然升至室温,搅拌反应48 h.所得反应溶液加入300 mL水,用二氯甲烷萃取,加入无水硫酸镁干燥,过滤.减压脱除溶剂,柱层析分离(洗脱剂:V(乙酸乙酯)∶V(石油醚)=1∶2),得淡黄色固体2.28 g,收率87.7%.1H NMR(500 MHz,CDCl3),δ:8.29(2H,Ar—H),4.12~4.08(8H,—CH2OOC—),3.77~3.75(4H,—NCH2—),3.30~3.27(4H,—CH2N3),2.63(8H,—OOC(CH2)2COO—),1.76~1.60(16H,—COO(CH2)4CH2N3),1.42~1.40(16H,—NCH2(CH2)4—).
化合物4的合成:
于1,6-己二醇(25 g,21.2 mmol)、2,3-二氢吡喃(8.9 g,10.6 mmol)的混合物中,加入对甲苯磺酸(0.012 5 g,0.725 mmol)和二氯甲烷(20 mL).室温反应24 h.反应完成后用饱和NaHCO3溶液洗除对甲苯磺酸,收集有机相,水相用二氯甲烷萃取,合并,用无水硫酸镁干燥,过滤.减压脱除溶剂,柱层析分离(洗脱剂:V(乙酸乙酯)∶V(石油醚)=4∶1),得浅黄色液体11.3 g,收率52%.1H NMR(500 MHz,CDCl3),δ:4.58~4.52(1H,—OCHO—),3.87~3.80(2H,—CH2CH2CH—),3.74~3.70(2H,—OCH2—),3.64~3.59(2H,—CH2OH),3.51~3.44(1H,—OH),3.42~3.33(2H,—CHOCH2—),2.13~2.07(2H,—OCH2CH2—),1.85~1.75(2H,—OCH2CH2CH2—),1.70~1.32(8H,—(CH2)4CH2OH).
化合物5的合成:

化合物6的合成:
化合物7的合成:
化合物M2的合成:
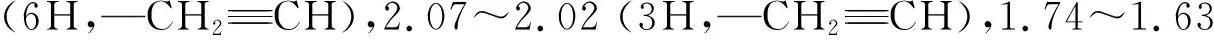
2.2 超支化聚合物hb-GPTA的合成
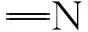
3 结果与讨论
3.1 单体及其中间体的合成
图1示出单体M1,M2及其中间体的合成.

图1 单体M1,M2及其中间体的合成
图1中:6-氯-1-己醇与叠氮化钠反应,得到化合物1,相应的核磁共振氢谱图2(a)中,δ3.3处叠氮亚甲基氢出峰化合物1.与丁二酸酐发生开环反应,引入羧基,得到化合物2,图2(b)中δ9.25处羧基出峰;化合物3由均苯四甲酸二酐和6-氨基-1-己醇反应得到,由于其溶解性差,并未测试其核磁谱图.化合物2与3发生酯化反应,产物在常见有机溶剂中具有良好的溶解性,在图2(c)中(δ8.29)苯环出峰.另外,δ4.12~4.08处为与酯基相邻的亚甲基上的氢,δ3.77~3.75处的三重峰为氮原子相邻的亚甲基上的氢,δ3.30~3.27处的三重峰属于与叠氮相邻的亚甲基上的氢,δ2.63处的单峰属于2个酯基之间的亚甲基,δ1.76~1.60,δ1.42~1.40处则为相同化学环境下的其他亚甲基上的氢,且各峰面积比例与氢原子个数比例一一对应,证明了单体M1成功合成.
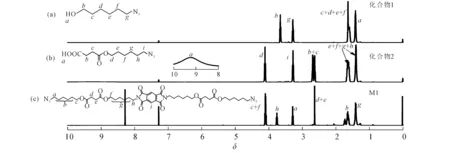
图2 化合物1,2和单体M1的核磁共振氢谱
1,6-己二醇与二氢吡喃在对甲苯磺酸的催化下,选择性保护一个羟基,在核磁共振氢谱图3(a)中,a位置的化学位移为δ3.42~3.33,2.13~2.07,1.85~1.75,3.87~3.80,4.58~4.52分别对应于环上的氢原子,且其峰面积之比与化学式相对应,符合化合物4的结构.保护后的产物4与溴丙炔反应,引入炔基,在核磁共振氢谱图3(b)中,δ2.4出现炔氢的特征峰,证明化合物5的成功合成;化合物5在浓盐酸的作用下,脱去保护基,得到羟基化合物6,相对于图3(b),环上氢的特征峰消失;该化合物与丁二酸酐反应,引入羧基,随后与三羟基乙烷发生酯化反应,得到单体M2,在图3(e)所示的核磁共振氢谱中,出现甲基特征峰.

图3 化合物4,5,6,7和单体M2的核磁共振氢谱
3.2 聚合物的合成与表征
图4为单体M1,M2和聚合物hb-GPTA的红外光谱,在3 300,2 100 cm-1分别是炔氢和叠氮的吸收峰,对比两个单体,聚合物在3 140 cm-1处出现了三氮唑基团的吸收峰,且3 300,2 100 cm-1的炔氢和叠氮的吸收峰明显减小,证明了点击反应的进行.运用核磁共振氢谱进一步分析,图5(a)为单体和聚合物的核磁共振氢谱,其中δ2.05为端炔氢的特征峰,δ3.30为叠氮亚甲基的特征峰,且其他峰的出峰位置及峰面积比例均正确,经过点击反应聚合得到聚合物后,聚合物的核磁共振氢谱显示在δ7.56和δ4.38~4.36分别出现三唑环上质子氢和连接在三唑氮原子上亚甲基氢的特征峰,δ1.98处为聚合物端炔氢的特征峰,进一步证明了聚合反应的进行.图5(b)为聚合物的核磁共振碳谱,通过核磁共振碳谱可知,δ145.37和δ122.13处存在三唑环上碳原子的特征峰,δ80.00存在炔碳的特征峰,由此也证明超支化聚合物的成功合成.

图4 单体M1,M2和聚合物hb-GPTA的红外光谱
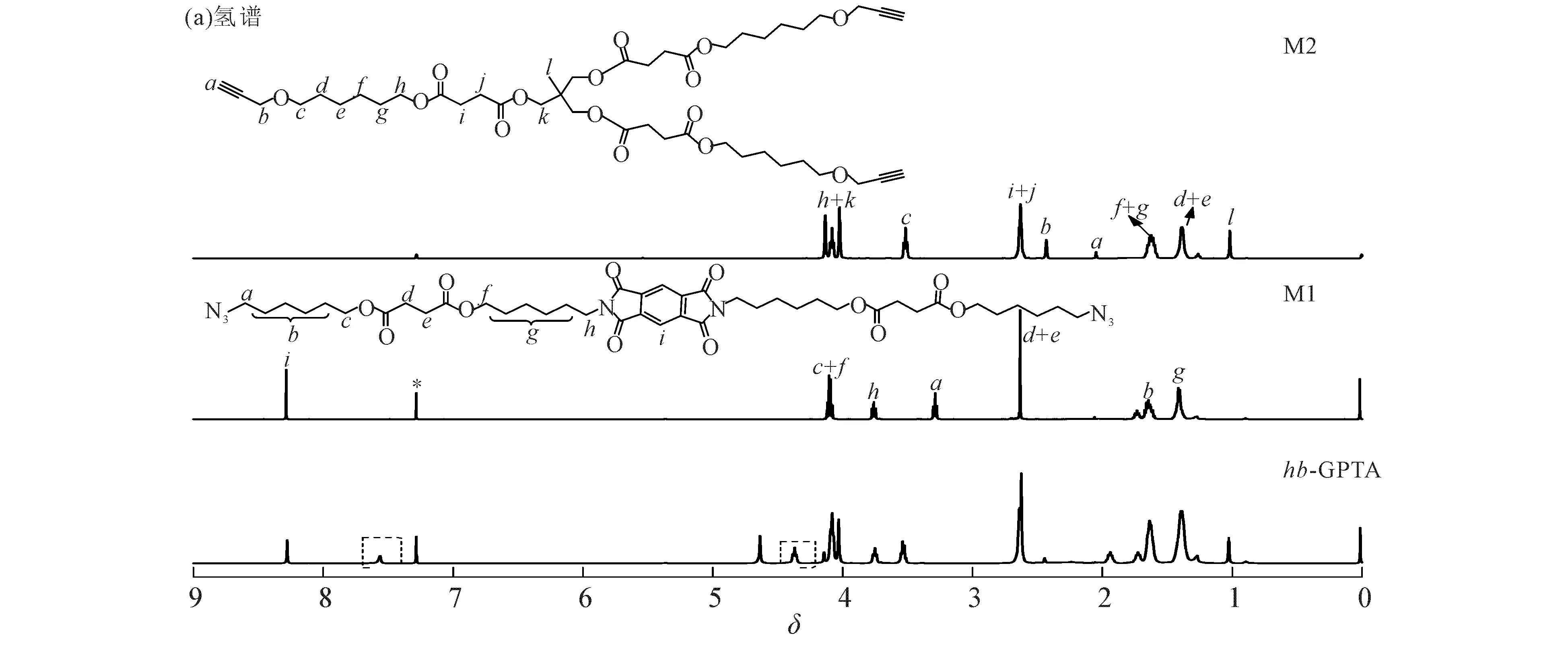
图5 单体M1,M2和聚合物hb-GPTA的核磁共振氢谱和碳谱
3.3 超支化聚合物的热稳定性及自修复性能
图6为聚合物的热失重曲线,以5%损失质量时的温度作为热分解温度,聚合物的热分解温度可达350 ℃,说明其具有良好的热稳定性.对聚合物自修复性能做了尝试,在室温以及60 ℃条件下未出现自修复现象,当温度提升至110 ℃,聚合物出现粘接现象,说明其在高温下具备一定的自修复性能,如图7所示.
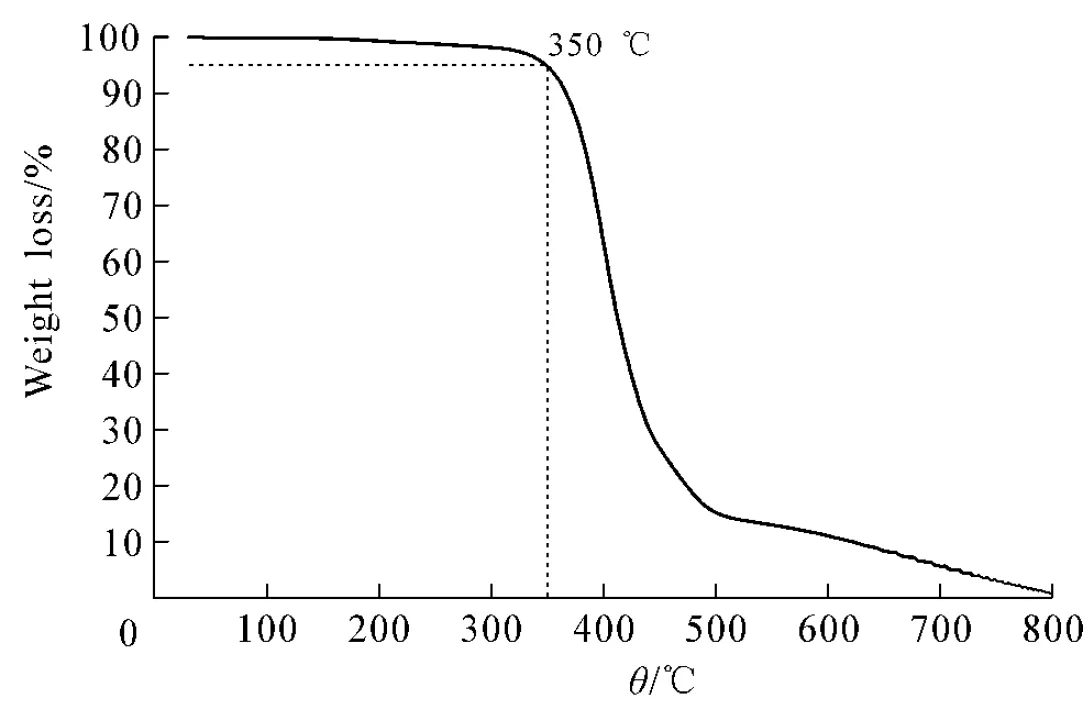
图6 hb-GPTA的热失重曲线

图7 hb-GPTA的自修复性能
3.4 超支化聚合物的成膜性
作为一类聚合物材料,可加工性是其能被加以应用重要前提.溶液成膜是制备聚合物薄膜的常用方法之一,将聚合物用二氯甲烷至刚好溶解,取少量溶液平铺在玻璃板,溶剂自然挥发,可得到一层透明、柔软的薄膜,成膜性较好(图8).

图8 超支化聚合物hb-GPTA的合成
3.5 超支化聚合物的导电性与电化学稳定性
通过溶液法,将超支化聚合物分别与LiTFSI和Zn(OTf)2按一定的质量比共混,得到相应的电解质材料.具体为:对于hb-GPTA/LiTFSI体系,比例为m=m(LiTFSI)∶m(hb-PTA)=0.8~2;对于hb-GPTA/Zn(OTf)2体系,m=m(Zn(OTf)2)∶m(hb-PTA)=0.8~1.8.测试不同比例下,室温电化学阻抗谱,经换算,得出电导率,结果如图9所示,hb-GPTA/LiTFSI体系的最高电导率为32.7 μS/cm(m=1.6),比文献值高了约一个数量级[7].对于hb-GPTA/Zn(OTf)2体系,最高电导率为0.42 μS/cm(n=1.2).

图9 hb-GPTA/LiTFSI和hb-GPTA/Zn(OTf)2体系的电导率随共混质量比的变化
通过线性扫描技术对材料的电化学窗口(ESW)进行表征.选取离子电导率最高的掺杂比体系,以不锈钢电极为正极,锂片或锌片为负极,利用线性扫描法测试材料的电化学窗口,结果如图10所示.LiTFSI掺杂时(hb-GPTA/LiTFSI),体系具有较宽的电化学窗口,其氧化还原的稳定范围为-0.7~+4.5 V,1.7 V左右的出峰可能是由于样品中残留的水份[14],因此其电化学窗口为5.2 V,与文献相当.Zn(OTf)2掺杂时(hb-GPTA/Zn(OTf)2),体系在整个扫描范围都保持稳定,因此,在相同条件下,聚合物电解质在锌体系下具有更好的电化学稳定性.
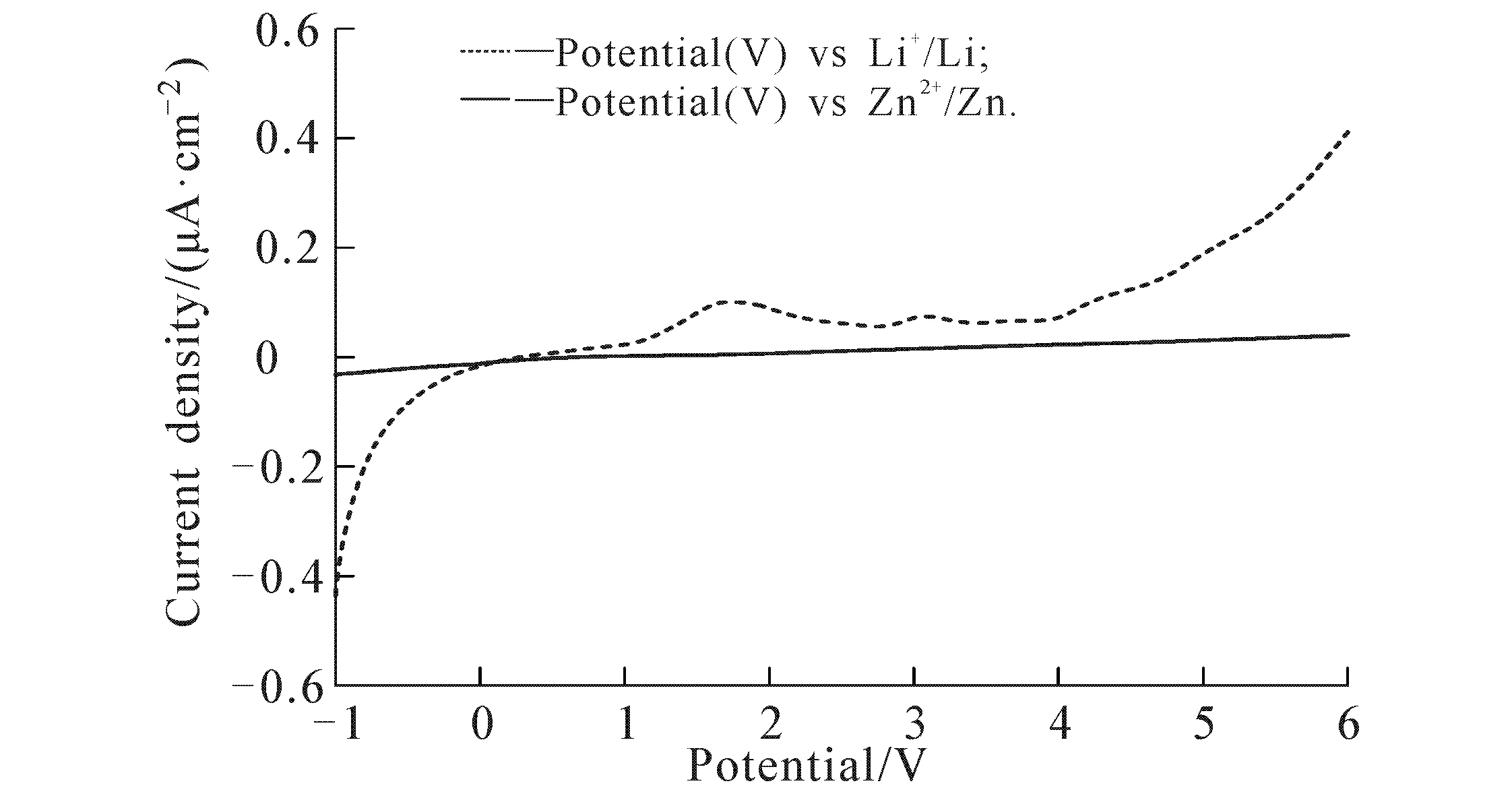
图10 hb-GPTA/LiTFSI和hb-GPTA/Zn(OTf)2的线性扫描伏安曲线
4 结论
基于叠氮-炔点击化学反应,制备了具有刚性结构的固体超支化聚合物hb-GPTA,运用红外光谱、核磁共振谱图对单体及聚合物的结构进行了表征,证明目标产物的成功制备;对聚合物进行了热失重分析,热分解温度可达350 ℃,热稳定性好,且聚合物具有较好的成膜性.以聚合物为基底与电解质盐进行共混,得到两种不同类型的固体聚合物电解质,当用LiTFSI掺杂时,最高离子电导率为3.27×10-5S/cm,电化学窗口为5.2 V,当用Zn(OTf)2掺杂时,最高离子电导率为4.20×10-7S/cm,且在-1~6 V电压范围内均能保持稳定.结合电化学稳定性分析,含三氮唑的聚合物在锌离子电池也具有潜在的应用前景.此外,本文得到的电解质电导率还有提升空间,如将分子结构中的烷基更换成烷氧或硅氧烷,或进行离子化反应,得到离子化的超支化聚三唑离子.
猜你喜欢
云南化工(2023年7期)2023-08-01 07:59:34
现代临床医学(2022年4期)2022-09-29 07:36:10
中国抗生素杂志(2022年7期)2022-08-18 03:22:36
商品与质量(2019年32期)2019-11-29 05:56:00
长春师范大学学报(2019年4期)2019-04-29 05:51:36
火工品(2018年1期)2018-05-03 02:27:56
中国资源综合利用(2017年3期)2018-01-22 02:45:40
中国果菜(2016年9期)2016-03-01 01:28:41
合成化学(2015年9期)2016-01-17 08:57:14
西华师范大学学报(自然科学版)(2015年3期)2015-02-27 15:31:19
