
糖尿病增加射血分数保留型心力衰竭的研究进展
2020-04-17 13:15唐晓笛
心肺血管病杂志 2020年2期
唐晓笛 樊 瑛
根据世界卫生组织最近的全球报告,糖尿病(diabetes mellitus,DM)人口自1980年以来翻了2倍,2014年达到4.22亿人[1],心血管疾病(CVD)仍然是糖尿病患者死亡的主要原因。世界上50%以上的糖尿病患者生活在亚洲,仅在东南亚就有超过7 200万的DM患者,并且患病人数仍然在快速增长[1]。从流行病学调查中获得的大量数据表明,射血分数保留的心力衰竭(heart failure with preserved ejection fraction,HFpEF)是DM患者的常见合并症,并且预示着不良的短期和长期预后[2]。据报道,HbAlc每升高1%,心力衰竭患者的再入院率和全因死亡风险就增加了25%[3]。DM不仅通过加速动脉粥样硬化,而且通过其他直接的细胞机制,对心肌细胞产生不利影响。
HFpEF是2018年中国心力衰竭诊断和治疗指南根据心力衰竭患者LVEF提出的一种新的分类方法。HFpEF是指具有心力衰竭的症状和/或体征,LVEF≥50%,BNP升高[BNP>35 ng/L或NT-pro BNP>125 ng/L],并符合以下至少1条:①左心室肥厚和/或左心房扩大,②心脏舒张功能异常。HFpEF是最常见的心力衰竭类型,其特点是具有心力衰竭的症状、体征和正常或接近正常的LVEF。目前此类患者病因、病理生理、治疗和预后尚不清楚[4]。很多文章报道了LVEF降低的HFrEF合并DM患者更高的再住院率及更长的住院时间,甚至研究了很多降血糖药物对HFrEF愈后的获益和风险,然而,关于HFpEF合并DM患者的临床特点、发病机制和治疗方法的相关研究却少之又少。
因此,我们迫切需要相关临床研究数据来理解合并DM的HFpEF的发病机制、临床特点、远期愈后和潜在的治疗靶点,以便更好的降低心力衰竭患者的再住院率和死亡率。本文就HFpEF合并DM患者的几种重要病理生理机制以及临床研究现状做一综述。
1.糖尿病增加射血分数保留型心力衰竭可能的病理生理机制
(1)糖脂代谢紊乱 人体中有两种主要供能方式,一种是血浆中葡萄糖通过葡萄糖转运蛋白4(glucose transporter4,GLUT-4)转运至细胞内进行有氧氧化功能,另一种方式则是血浆中游离脂肪酸(fatty acid,FA)通过跨膜转运至细胞内参与β氧化供能。研究发现,过氧化物酶体增殖物激活受体(Peroxisome proliferator activated receptorα,PPARα)可能是调节两种代谢平衡的关键靶点,PPARα是一种由配体激活的转录因子受体,属于核受体超家族成员,主要表达于心脏、肝脏及骨骼肌系统,PPARα激活可能促进脂质β氧化代谢[5]。众所周知,心肌细胞60%~70% ATP是由FA的β氧化提供的,其余部分则由葡萄糖、酮体等其他物质提供,并且心肌细胞的能量摄取受胞浆中两种物质的浓度调节。在糖代谢过程中,细胞中葡萄糖跨膜转运由GLUT-4介导,DM时由于胰岛素抵抗或缺乏,细胞膜表面的GLUT-4表达下调[6],心肌细胞葡萄糖的摄取和氧化受限,导致葡萄糖利用障碍。GLUT-4表达下调会诱导心肌细胞膜过表达CD36(FA转位酶)和FA结合蛋白(fatty acid binding protein,FABP)[7],介导FA进入胞内,同时脂肪酸β氧化限速酶CPT-1表达上调,促进FA转运入心肌细胞线粒体内进行β氧化[8]。FA摄取和氧化不平衡会导致脂质在细胞内过度积累[9],抑制PPARα活性,进一步造成FA利用障碍,形成恶性循环。糖代谢和脂代谢障碍使心肌细胞ATP产生减少,同时游离FA的累积又加重胰岛素抵抗,并产生心肌毒性脂类物质如神经酰胺和二酰基甘油,而脂毒性会导致线粒体功能障碍并促进细胞凋亡,形成心肌细胞纤维化和心室重构,加快心力衰竭的进展。FA和葡萄糖氧化利用均减少,ATP产生不足,这种低效率的能量供给进一步加重心肌重构,而这种能量供应障碍,首先影响的就是心肌细胞的舒张功能,这或许是DM常合并HFpEF并且病情进展迅速的可能原因。心力衰竭终末期,线粒体氧化磷酸化解偶联,心肌处于严重的“能量饥饿”状态[10],药物治疗已经收效甚微,等待心脏移植可能是其唯一的治疗方法(图1)。
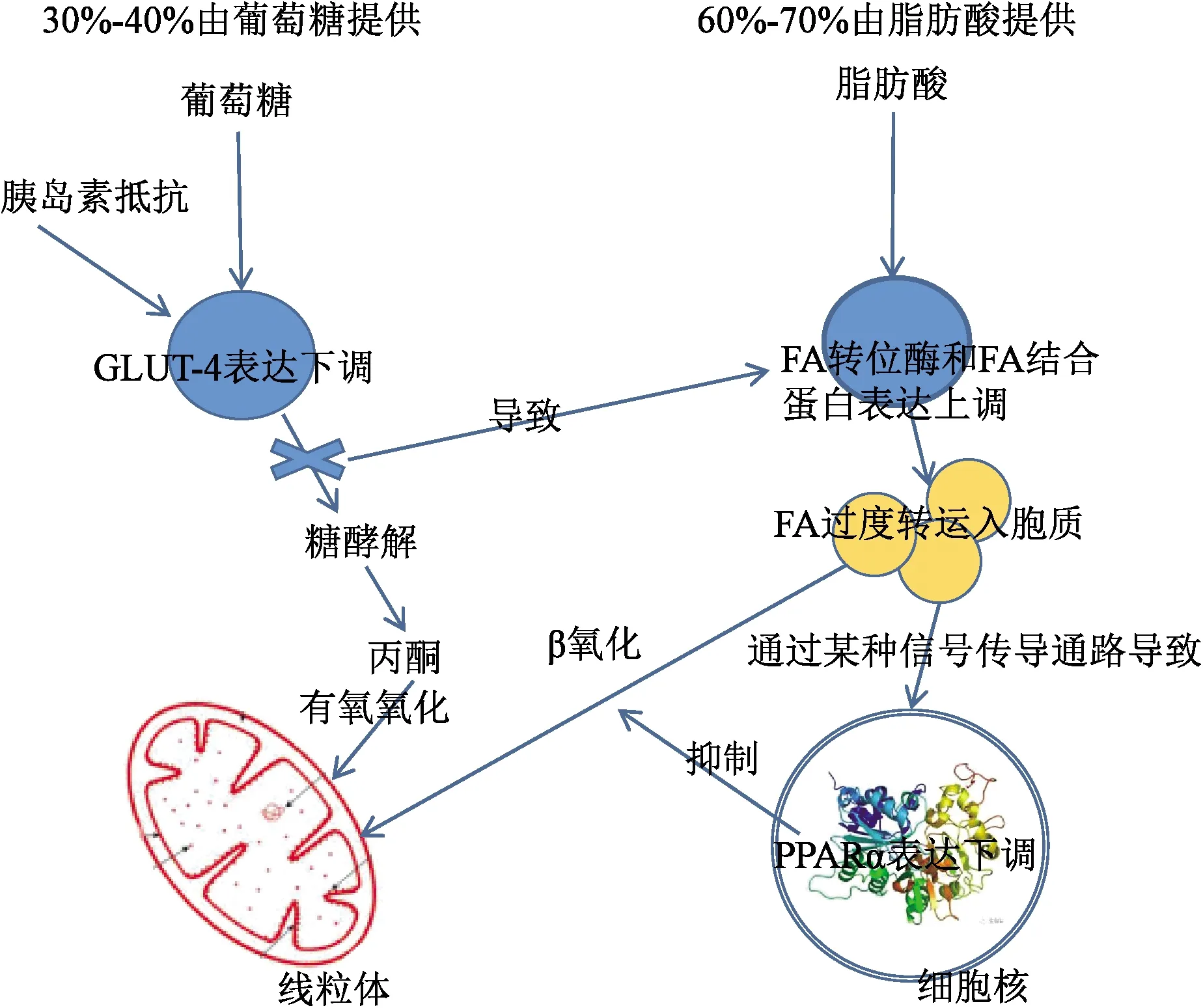
图1 糖脂代谢路线图
(2)钠-葡萄糖协同转运蛋白-2(sodium-glucose cotransporter-2,SGLT-2)表达上调 长期过高的心室负荷是诱发心力衰竭的重要病理生理机制。DM患者升高的血浆葡萄糖浓度可能通过交感神经和肾素-血管紧张素系统的过度激活引起SGLT-2表达上调,导致肾小管和集合管对钠水重吸收增加,对利尿剂反应性下降,造成液体潴留,使心脏负担加重,增加心力衰竭的发病率和死亡率[11],但其中具体的病理生理机制还需做进一步研究。
(3)氧化应激、炎症标志物和晚期糖基化终末产物增多
胰岛素抵抗、血浆葡萄糖水平升高及代谢综合征通过一系列分子及细胞机制导致蛋白质、脂类和核酸的非酶糖化导致晚期糖基化终产物的形成而引发炎症反应和氧化应激,导致线粒体功能障碍,进而导致细胞凋亡和纤维化[12]。氧化应激是由于活性氧(ROS)生成增加和(或)抗氧化防御能力降低而引起的不平衡,这两种机制都会导致糖尿病心肌病[13],与高血糖相关的氧化应激被认为在糖尿病心脏并发症中发挥着关键作用[14],包括心脏重构,心肌细胞钙转运受损,心脏收缩和舒张功能降低,特别是心脏舒张功能受损[15],氧化应激通过激活心脏损伤的下游通路即炎症细胞因子的释放、巨噬细胞的聚集浸润而引发的一系列炎症反应,所有这些反应都可能导致线粒体氧化呼吸链功能障碍,ATP产生减少,活性氧增多,诱发更重的炎症反应,形成恶性循环,最终加重心肌细胞的结构变化、内皮功能障碍和多器官损害[11,16]。
(4)交感神经系统过度激活 血糖增高和胰岛素抵抗很可能受到自主神经系统功能紊乱的影响,DM患者可能有更高的交感神经活性[17]。血浆中高胰岛素负载可能是引发这一机制的潜在原因,高胰岛素血症通过增加磷酸酶4D(phosphodiesterase4D,PDE4D)表达而导致PKA通路过度激活而诱导交感神经兴奋性增高。王庆通、刘永明等通过构建糖尿病小鼠模型发现抑制胰岛素介导的β肾上腺素能活化可预防糖尿病相关的心脏收缩和舒张功能障碍,目前这一机制在人类身上还没有得到确切证实[18]。交感神经系统的过度激活可导致HF患者死亡率增加,因此,DM可能是心力衰竭的诱发因素之一。
(5)微循环障碍及心肌毛细血管减少致心肌舒张功能障碍 DM患者心肌微循环退化,毛细血管网变得更加稀疏,这直接导致了心室肌顺应性下降,心肌变得僵硬,舒张功能发生障碍。Rabea等[21]利用接受心脏移植患者(合并DM和未合并DM)终末期心脏进行了组织学分析发现,DM患者心肌毛细血管密度下降,同时伴随着血管紧张素II浓度升高。随后他们在胰岛素依赖性DM的转基因猪模型的心脏上发现了同样的病理改变,微血管失稳被认为是血管紧张素II过度表达造成的[19]。同时他们也发现胰岛素依赖性DM的转基因猪模型中THP-1细胞与血管内皮细胞粘附力增强,提示内皮细胞处于慢性炎症反应状态,然而过度的炎症激活和血管紧张素II过表达是否相关,还需要进一步研究。微循环退化被认为是大多数器官血管退化的原因,所有这些都证实了DM患者心脏微血管紊乱与心脏功能障碍具有相关性。接下来,实验人员继续使用两种众所周知的血管生成刺激物血管内皮生长因子-A(vascular endothelial growth factor A,VEGF-A)和磺胺噻二唑(Tβ4)[20]诱导微血管内皮细胞的形成,结果显示在正常和高糖条件下,Tβ4在体外均能增强内皮细胞的迁移能力,促进血管新生,但在高糖培养条件下作用较小,而VEGF-A仅能在正常条件下促进毛细血管新生,提示磺胺噻二唑(Tβ4)可能作为一种有效的治疗手段改善HFpEF合并DM患者愈后。同时,在DM的转基因猪模型上发现在高血糖条件下miR 92a、 miR 26a和miR133b表达明显增加,miR表达上调可能和微循环退化相关,未来可能通过抑制miR表达也能改善HFpEF合并DM患者微循环退化,延缓舒张功能障碍的进展[21]。
2.糖尿病增加射血分数保留型心力衰竭相关临床研究进展
有研究发现,约45%的HFpEF患者合并DM,并且在新发HFpEF的患者中合并糖尿病的比率在进一步上升。这就给我们一个启示,HFpEF可能并不是一个独立的疾病,而是多种潜在机制紊乱的外在表象,了解其相关临床研究现状,或许能更好的认识并改善其愈后[22]。
(1)关于糖脂代谢紊乱 PPARα作为糖脂代谢平衡的重要靶点,通过激活该转录因子受体,促进FA在线粒体内进行β氧化,减轻DM造成的FA摄取和氧化不平衡导致的脂质在细胞内过度积累,增加脂代谢途径ATP的生成,来改善心肌细胞能量供应状态,减少心肌细胞凋亡,减轻心肌细胞纤维化和心室重构的发生可能为一种有效的治疗手段。目前选择性PPARα激动剂尚处于大规模人群实验中,其作用机制还不是完全清楚,PPARα在调节糖脂能量代谢及其在糖尿病和心力衰竭中发挥的作用还需进一步研究。K-877 为一种新型 PPARα激动剂,二期临床试验证实,其可通过提高FGF21水平促进FA氧化功能,改善心肌细胞特别在合并糖代谢异常的情况下的能量代谢,有望于改善HFpEF合并DM的愈后[23]。
(2)关于SGLT-2表达上调 2017年I-PRESERVE实验观察发现,和无DM的HFpEF相比,合并DM的患者有更大的心室负荷和更高的NT-proBNP水平[24]。同样,由Crystel等观察研究的亚洲人群HF合并DM的患者有更高的左心室收缩压、舒张压和NT-proBNP水平及更糟糕的左心室收缩功能[25]。上述实验可能从流行病学观察上印证了升高的血浆葡萄糖浓度可能通过某种信号传导通路引起钠-葡萄糖协同转运蛋白-2表达上调从而导致液体潴留而加重HF。
关于降糖药物SGLT-2抑制剂的多个大型回顾性研究,例如纳入了上万人的大规模研究SGLT-2抑制剂心血管获益的EASEL、DECLARE-TIMI 58及EMPA-REG OUTCOME实验,它们都得出一致结论,使用SGLT-2抑制剂控制血糖可以降低心力衰竭的发生率和已经发生HFpEF患者的再住院率和死亡率,缩短HFpEF患者的住院时间,改善长期愈后[26-28],SGLT-2未来有望成为HFpEF合并DM患者的必备治疗手段,甚至已有将SGLT-2用于HF未合并DM患者治疗的临床研究。
(3)关于氧化应激、炎症标志物和晚期糖基化终末产物增多 在RELAX和I-PRESERVE实验中超声心动图结果显示:DM与严重的舒张功能障碍和左心室肥大密切相关,氧化应激、炎症反应和晚期糖基化终产物增多等相应代谢综合征可能是其相关诱因[11,16]。
针对氧化应激、炎症反应和晚期糖基化终产物增多等相应代谢综合征最有效的治疗手段似乎是减重和控制血糖,也有一些药物在降低血糖的同时能够抑制炎症反应和心室重构,例如胰高血糖素样肽-1受体激动剂(glucagon-like peptide-1 receptor agonists,GLP-1)、钠-葡萄糖协同转运蛋白-2(sodium-glucose cotransporter2,SGLT-2),这些药物能够改善内皮细胞功能紊乱和心室功能、延缓多器官损害。另外,沙库巴曲缬沙坦钠(neprilysin,sacubitril/valsartan)可能能够通过抑制利钠肽降解,使cGMP水平升高,导致蛋白激酶G的活化增加,改善内皮功能,降低心肌细胞僵硬度,目前相关研究正在进行中,结果令人期待[11]。
(4)关于交感神经系统过度激活 Witte等[29]对HF合并DM和未合并DM患者使用β受体阻滞剂和ACEI/ARB类药物,经过五年的随访观察得出这样的结论:高剂量的β受体阻滞剂会降低HF患者的死亡率,但合并糖尿病患者可能从高剂量的β受体阻滞剂中获得更多的好处,随后他们通过测定两组患者的肌肉交感神经活性,提出合并DM患者较无DM患者可能有更高的基础交感神经活性,通过这一研究结果结果我们很容易解释β受体阻滞剂剂量与糖尿病患者预后之间的更强联系与过度交感神经激活有关。
HFpEF合并DM患者较HF未合并DM患者可能有更高的拟交感活性,那么在临床治疗中增加β受体阻滞剂剂量或许更能改善愈后,但未来还需更多的大规模临床实验来支持这一结论。
(5)关于微循环障碍及心肌毛细血管减少致心肌舒张功能障碍 在动物实验中,Tβ4被证实可以促进高血糖状态下心脏毛细血管的新生,同时发现miR表达上调可能和微循环退化相关,那么未来通过外源性补充Tβ4或抑制miR表达来改善HFpEF合并DM患者微循环退化的临床研究值得期待。
总之,HFpEF可能并不是一种独立的心脏舒张功能障碍性疾病,它可能是多器官系统紊乱出现的心脏并发症,合并DM的患者在HFpEF中占大部分,同时这部分患者也具有更差的愈后,更高的再住院率及更长的住院时间,目前两种合并症的临床特点、发病机制还不十分清楚,国内外指南也没有提出具有明显获益的治疗手段,未来还需要多学科专家共同努力,探索这种共患病的发病机制,制定具有针对性的治疗手段,改善长期愈后,降低病死率。
猜你喜欢
现代临床医学(2022年4期)2022-09-29
中老年保健(2022年4期)2022-08-22
承德医学院学报(2022年2期)2022-05-23
昆明医科大学学报(2022年2期)2022-03-29
华中科技大学学报(医学版)(2021年6期)2022-01-16
保健与生活(2021年4期)2021-02-22
家庭百事通·健康一点通(2020年11期)2020-11-30
体育科学(2018年12期)2019-01-04
科学中国人(2016年9期)2016-11-04
哈尔滨理工大学学报(2014年3期)2015-01-04
