
Mutator超家族转座子研究进展
2020-04-13 06:29:28从春生李玉斌
遗传 2020年2期
从春生,李玉斌,2
综 述
超家族转座子研究进展
从春生1,李玉斌1,2
1. 中国农业科学院生物技术研究所,北京 100081 2. 青岛农业大学农学院,青岛 266109
转座子是一类可以在基因组中不同遗传位点间移动的DNA序列,在其转移过程中有时会伴随自身拷贝数的增加。作为基因组的重要组成部分,转座子可以通过多种方式影响宿主基因及基因组的结构与功能,进而在宿主的演化过程中扮演重要角色。目前依据转座过程中间体类型的不同可以将其分为I类转座子和II类转座子。超家族转座子是20世纪70年代在玉米(L.)中发现的一类特殊的转座子,其属于II类转座子,广泛存在于真核生物基因组中,包含遗传特征明晰可分的众多转座子家族。此外,该超家族转座子转座频率高,倾向于插入基因富含区及低拷贝序列区,可快速产生大量新的突变体,目前已被广泛应用于正向及反向遗传学研究。本文结合近年来相关研究结果,围绕超家族转座子的分类组成、结构特征、转座机制、插入偏好、靶位点重复序列以及玉米自主性元件展开综述,并对转座子研究面临的问题及未来研究方向进行了探讨,旨在与研究领域内的同行探讨相关研究的可能突破点、未来发展方向及可能产生的重大影响。
超家族;家族;元件;转座机制;插入偏好;靶位点重复序列
20世纪40年代美国遗传学家Barbara McClintock在玉米中发现了一些在染色体上可以移动遗传位置的元件,并于50年代初提出可移动的遗传基因(即“跳跃基因”)学说,但直到30年后这一超越时代的学说才被科学界同行逐渐理解和接受,并将这些可以在基因组中移动的DNA序列统称为转座元件或转座子[1]。现有研究表明,转座子几乎存在于所有生物基因组中,是基因组的主要组成部分。由于其重复特性,转座子曾一度被认为是垃圾DNA,但越来越多的证据表明转座子是塑造基因组的重要因素[2~5]。在许多植物基因组中,半数以上的序列属于转座子[6~12],特别在玉米基因组中转座子序列的占比更高达85%[11],并且这些转座子序列通过不同方式影响了玉米的驯化[13]、传播[14,15]及优异农艺性状的形成[16,17]等。此外,转座子在自身序列、蛋白功能域和结构等方面具有十分丰富的变异[18]。按照转座过程中间体的类型,真核生物转座子可以划分为两大类:Ⅰ类转座子(RNA转座子或反转录转座子)和Ⅱ类转座子(DNA转座子)[19]。Ⅰ类转座子的转座反应通过DNA-RNA-DNA形式介导完成,通常以“copy- and-paste”方式进行转座,按照其长末端重复序列(long terminal repeats, LTR)的有无又可以分为LTR反转录转座子和non-LTR反转录转座子2个亚类;Ⅱ类转座子的转座反应通过DNA-DNA形式介导完成,绝大部分以“cut-and-paste”方式进行转座,少数转座子通过滚环模型或自我合成途径完成转座反应[20,21]。根据序列组成和基因结构特征,Ⅰ类和Ⅱ类转座子又都可分成不同的超家族[22],各超家族间既存在共性,同时又各有特性,尽管采用的分析方法不尽相同[23~25]。超家族转座子属于Ⅱ类转座子,广泛存在于真核生物基因组中,包含着遗传特征明晰可分的众多转座子家族,并且在转座子遗传特性方面的研究深入,在功能组学研究中的应用也十分广泛。本文结合近年来的研究结果,围绕超家族转座子的分类组成、结构特征、转座机制、插入偏好、靶位点重复序列以及玉米自主性元件进行了概述,同时对转座子研究面临的问题及未来研究方向进行了探讨,以便相关科研人员更充分、全面了解超家族转座子的研究进展。
1 Mutator超家族转座子分类组成
1978年,美国爱荷华州立大学(Iowa State University)的Donald Robertson博士报道了一份高突变频率玉米材料,其幼苗中的突变频率接近自发突变的30倍左右,而这一突变特性的遗传不符合经典的孟德尔遗传定律[26]并表现出明显的表观沉默[27]。这一遗传品系由于存在大量一类新型转座元件—或[28~30],从而可以发生高频突变,也因此被称之为系[31]。目前这些转座元件同属家族或家族,其中可以编码转座酶并使其自身发生转座的元件称为自主性转座子—(-Donald Robertson),而在活性存在时才能进行转座的元件统称为非自主性转座子[32~34]。与大量的非自主性转座子组合可形成高效的突变系统,系统是目前被广泛应用的致变能力极强的转座子插入突变体创制系统[35~37]。
另外,minimal系是通过筛选获得的只含有单一和一个位于颜色基因中的非自主性的遗传品系,成为研究玉米转座子系统及调控的理想材料[38~40]。例如,从minimal系中发现了()[41]。作为调控的显性遗传性位点,可以沉默一个或多个活性,但并不是维持沉默状态所必需的,在后代分离个体中,即使缺失位点,仍无活性并且可以维持多代[42]。的发现极大地促进了转座子表观沉默的研究,同时使用于突变体创制的系统变得更为可控。
近年来,伴随着测序技术的发展以及被测序物种数量的不断增加,在植物[43~47]、真菌[48,49]、原生动物[50,51]以及多细胞动物[52,53]中均发现了与玉米序列相类似的转座子,统称为元件(-like transposable elements)。目前大部分鉴定出来的元件都属于非自主性转座子,它们自身不能编码功能完善的转座酶,只有极少数元件可以进行自主转座,例如尖孢镰刀菌()中的[49],拟南芥()中的[54],玉米中的[55]和[56],水稻()中的[57]以及埃及伊蚊()中的[58]等。另外,大量非自主性元件内部有时携带着来源于宿主的一个或多个不同基因的片段,这类元件被特别命名为Pack-MULEs。目前在拟南芥、水稻、玉米、百脉根()、西红柿()及荷花()的基因组中都发现了Pack-MULEs的存在[59~65],其中水稻中Pack- MULEs的数量巨大,有关研究也更为深入。水稻中有些Pack-MULEs元件所携带的多个宿主基因片段可形成崭新的开放阅读框并转录出嵌合转录本。氨基酸序列功能分析及蛋白组学研究表明,捕获的基因片段甚至可能具有特定的功能。结合以上研究结果及Pack-MULEs在植物中的普遍性,Jiang等[63]推想Pack-MULEs获取基因片段的方式很可能是高等植株基因进化的一种重要机制。虽然Pack-MULEs捕获宿主基因组片段的分子机制目前仍不清楚,但研究发现Pack-MULEs主要倾向于获得和保留GC含量高的序列,这种选择性捕获使Pack-MULEs更有可能捕获具有功能性的序列,进而为新基因的进化及现有基因的修饰提供新的遗传资源[66~68]。与此同时,相对于其他超家族转座元件,水稻中Pack- MULEs表现出独特的表观遗传学特性,其插入和表达不仅可以改变水稻染色体的表达模式,还可以抵消重组对染色体碱基组成的影响,进而对染色体结构进化产生影响[69]。
2 Mutator超家族转座子及其转座酶基本特征
与其他大多数DNA超家族转座子相比,超家族转座子两端具有较长的末端反向重复序列(terminal inverted repeats, TIR)。TIR序列中包含有转座酶结合位点[70],而携带单一TIR的转座子无法正常进行转座[60,71]。此外,TIR序列中还含有复杂的启动子序列,既可以启动转座酶或TIR间序列的转录,也可以调控转座酶在不同组织中的表达[63,72]。玉米家族转座子的TIR比较保守,大多长约215 bp[73],根据两端TIR间序列的差异,又划分为不同亚家族(~)[74~76]。其中大部分为非自主性转座子,这些非自主性转座子是内部片段缺失产生的衍生物或者是其他序列点突变导致转座酶功能丧失的同源序列(homologs, h)。h虽然不能催化转座反应,但可能在表观沉默中发挥增强作用[31]。相对于玉米家族转座子,各种元件TIR序列变异丰富。有些元件TIR内含有串联重复序列,这些串联重复序列可能导致TIR自身形成特殊的二级结构,进而影响转座子的转座行为[77]。在植物和真菌中大部分元件具有较长的TIR (100~ 600 bp),但在拟南芥[65]、荷花[61]、玉米[78]和酵母()[48]基因组中鉴定到少数non-TIR元件(TIR<50 bp),这些元件虽分布较为广泛,但其与元件在进化中的关系仍不清楚。另外,在玉米、西红柿、水稻和拟南芥基因组中还检测到一些多TIR元件,这些TIR大多以串联形式分布,多TIR元件可能更有利于转座子转座和捕获宿主基因组序列[60]。
作为家族中的自主性转座子,同时也是超家族转座子研究的典型代表。编码两个转录方向相向的基因:和,各自转录起始于两端的TIR内部序列,两个转录本间没有重叠部分,在相距200 bp处终止转录[32](图1A)。编码蛋白MURA (94 kDa),MURA与原核生物转座子的转座酶序列相似[79],含有保守的蛋白结构域[80],被认为是转座酶,催化转座子转座。编码蛋白MURB (23 kDa),MURB并非体细胞组织转座剪切所必需,可能与生殖类细胞内转座子的重新插入相关[81,82],MURB调控方式及其在转座过程中的功能目前还没有更为详尽的报道。如前所述,在玉米及其他植物、动物、微生物中也已经鉴定到了少数几个自主性元件,但这些新发现的自主性元件均只含有同源基因,因此基因可能仅存在于玉米的中。是转座插入并重排形成的2.2 kb反向重复序列,由两段反向加倍的TIRA及其下游相邻部分序列组成,不涉及任何基因序列(图1B)。插入位点两翼残存的两个转录方向相向的基因(和)中仅有启动子起始转录,由此产生的发卡状转录本生成小RNA (主要是22 nt siRNAs),然后通过RNA介导的DNA甲基化方式沉默活性[41,42]。
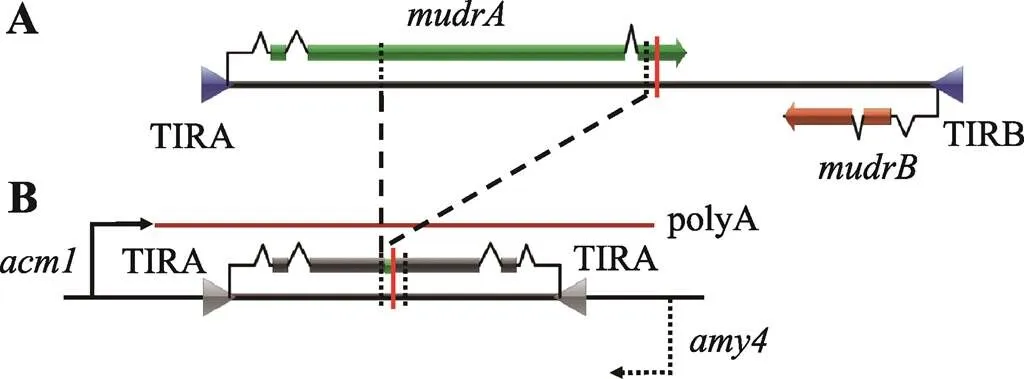
图1 MuDR和Muk基因结构组成
A:转座子及其基因和的结构;B:及其两翼残存基因的结构。
转座酶是自然界中最丰富、最普遍存在的基因编码产物[83]。所有真核生物“cut-and-paste”类型超家族转座子其转座酶均具有DDE/D三氨基酸特征结构域[84],及其他自主性元件转座酶同样具有这样的特征(图2)。Liu等[58]根据保守的DDE/D结构域并通过生物信息学方法在埃及伊蚊基因组中发现了自主性元件—,通过定点突变首次证实了元件转座酶中DDE/D结构域3个特征氨基酸的重要性:其中任何单一氨基酸的改变都足以使转座酶的活性完全丧失。此外,与另一类DNA转座子超家族—超家族的转座酶类似,大部分超家族成员的转座酶在DDE结构域的第2个D和E之间还含有一个保守的CXXH基序和一个色氨酸[85](图2)。CXXH基序可能参与转座酶对TIR的识别,当CXXH基序中的组氨酸突变后,转座酶催化活性消失殆尽[58,86]。而色氨酸不仅与转座酶活性相关,还与转座酶的精确切割或修复相关。当转座酶中色氨酸突变为丙氨酸时,转座酶催化活性彻底消失;当其突变为其他芳香族氨基酸时,转座酶表现出一定活性,但转座子剪切频率变低,精确剪切比例也显著下降[58,85]。
除了DDE/D三氨基酸这一特征结构域以外,在超家族转座子的转座酶中还可以鉴定到其他保守结构域(图3A)。例如,大部分转座酶的N端具有属于WRKY-GCM1超家族[87]的DNA结合结构域(DNA binding domain, DBD),可能通过结合转座子特定区段序列来调控转座酶活性及转座子的转座行为。水稻编码的转座酶DBD上游特定长度编码序列发生缺失突变后,转座子的剪切频率显著增高,进一步研究发现这部分片段中氨基酸组合的理化特性对转座酶活性至关重要[71]。另外,大量MURA同源蛋白C端也具有相对保守的基序,如在拟南芥、玉米、水稻和甘蔗(spp.)中先后鉴定到CX2CX4HX4 (或6) C基序[47,65],目前已知的自主性超家族转座子的C端 大多存在这些锌指基序(图3B),它们可能通过结 合核酸序列(DNA或RNA)参与调控转座子的转座行为。

图2 不同自主性转座酶DDE结构域蛋白序列比对分析
黑色阴影表示氨基酸完全一致,粉色阴影表示同源性≥75%,绿色阴影表示同源性≥50%,保守的氨基酸及结构标注在序列底部。
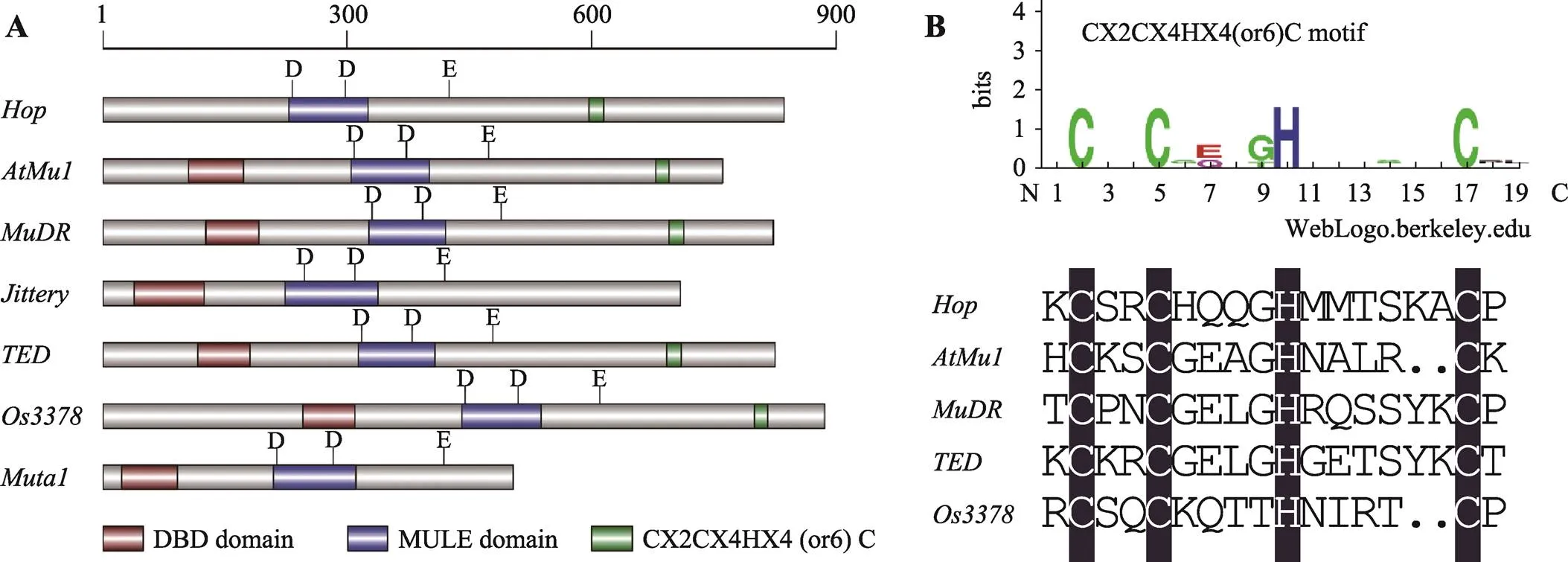
图3 Mutator超家族转座酶的结构特征
A:及其他自主性元件转座酶的保守结构域;B:超家族转座酶C端的保守基序。
3 Mutator超家族转座子转座机制
利用家族转座子特有的遗传组成和转座特性,已经构建了多个玉米突变体资源库(如TUSC、MTM、RescueMu、UniformMu和ChinaMu等),为正向遗传学和反向遗传学的研究提供了丰富的突变体遗传材料[88,89]。然而,关于超家族转座子转座机制的认识仍缺乏直接的证据。基于与其他超家族DNA转座子的一些共性及大量转座事件分析,推测超家族转座子的剪切及再次插入可能与某些已知的转座机制存在相似之处。
3.1 转座酶催化作用下的DNA双链断裂
真核生物DNA转座子的剪切过程一般以转座子两端某一条DNA单链的解离为起始,该过程为亲核裂解反应,通常H2O作为亲核试剂,在转座酶的作用下攻击转座子与侧翼序列连接处的磷酸二酯键而形成断裂口。某些超家族转座子会在转座子末端暴露出自由的3′-OH,而其他超家族转座子则在侧翼宿主序列末端暴露出自由的3′-OH,随后,不同类型超家族转座酶催化不同位置的3′-OH与不同类型DNA底物组合而使第二链断开,由此形成的DNA双链断裂(DNA double strand break, DSB)使转座子最终得以从供体位点释放出来[90]。最近研究发现,埃及伊蚊第二链断开方式与、超家族转座子相类似[77]。在转座酶作用下,以H2O作为亲核试剂使转座子末端与侧翼DNA连接处的磷酸二酯键断开后在侧翼DNA的3′末端暴露出羟基,3′-OH进攻另一链而在侧翼DNA末端形成发卡结构,最后释放出转座子(图4)。剪切位点形成的DSB既可以通过非同源末端连接(non-homologous end joining, NHEJ)方式修复,留下不同类型转座印迹(footprint),还可能通过同源重组(homologous recombination, HR)方式,以一条姐妹染色单体或同系物作为模板进行修复。但是,侧翼DNA形成的发卡结构必须在修复前打开,相关的体外实验表明,这一过程并不是由转座酶催化完成,而可能是由宿主自身可以切割类似发卡结构的酶来完成[86]。目前对于超家族转座酶催化作用下的DNA双链断裂过程报道较少,该过程是否是超家族转座子的共同遗传特性亟待其他自主性超家族转座子相关研究加以验证。
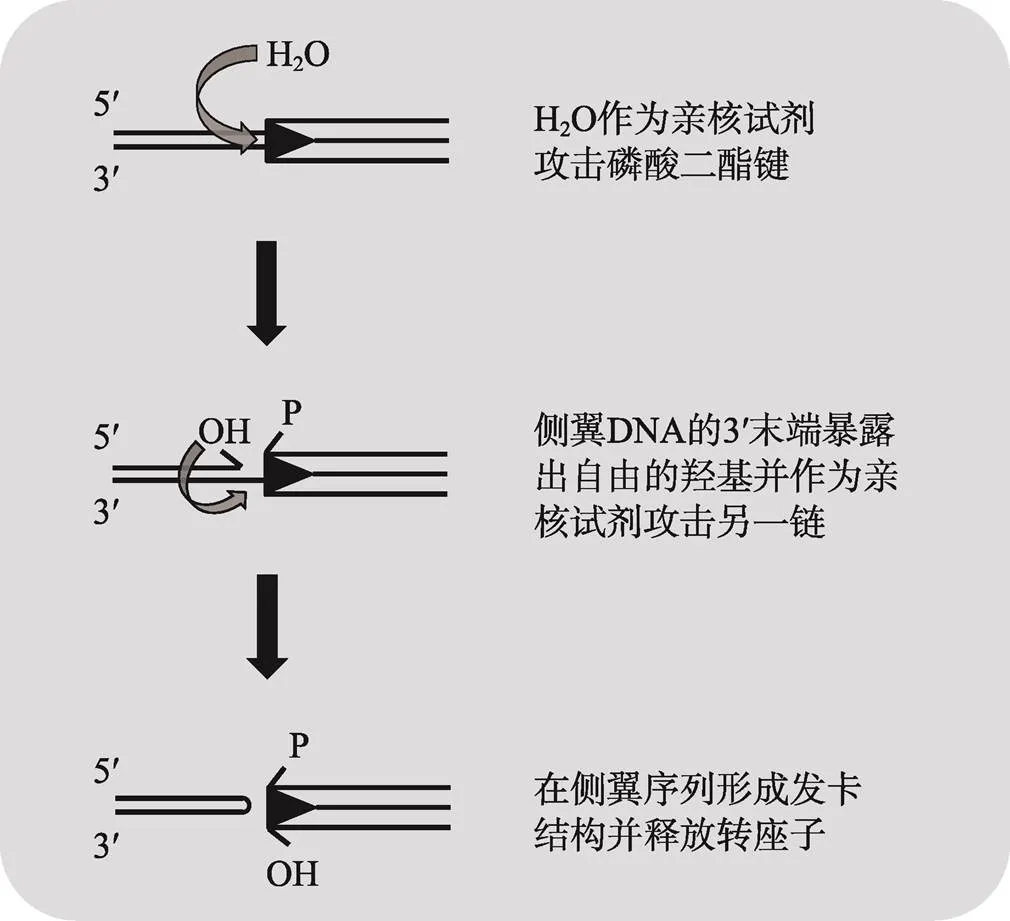
图4 Muta1转座酶介导的DNA双链断裂过程
3.2 转座子剪切后的DSB修复
转座子剪切后导致DNA发生双链断裂,目前认为植物不同组织细胞中DSB修复方式不尽相同[91,92](图5)。若剪切发生在生殖类细胞(包括配子体及配子体减数分裂前的有丝分裂细胞)的S期或G2期,此时转座子已经随着染色体发生了复制,细胞能够以姐妹染色单体为模板进行精确修复。由于剪切位点被完全修复,转座反应看似以“copy-and-paste”方式进行,但事实上是转座子剪切后又被重新修复的结果,而并非转座子的简单加倍。在DSB的修复过程中,由于模板内存在一些长短不一、散落分布的微同源序列,修复复制链发生位置滑移,便形成了大小有别、序列组成不同的多种缺陷型转座子。而在体细胞发育过程晚期,转座子剪切可能发生在细胞S期之前或者剪切形成的DSB主要通过易错易突变的NHEJ方式进行修复,结果导致转座子原插入位点产生多种类型的footprint序列[93,94]。
目前已有多方面证据支持上述假说。在和生殖类细胞转座研究中均可以检测到缺陷型转座子,并且这些缺陷型转座子在缺失序列的两翼存在微同源序列[56,91]。此外,研究还发现在一些缺陷型转座子序列内部含有填充序列(filler DNA),并且这些filler DNA均来自缺失位点附近序列[56,95]。这正是由于修复模板内存在多组微同源序列,修复复制时发生了多次复制链滑移造成的。由此可见,在生殖类细胞中转座子剪切后是通过依赖模板的方式进行修复的。另外,研究发现在玉米RAD51突变体中生殖类细胞内转座子转座行为异常。RAD51在细胞减数分裂的DSB修复过程发挥重要作用,玉米中存在两个同源基因,在含有活性的RAD51双突变材料中,生殖细胞转座反应中内部及侧翼序列缺失的频率比野生型材料高出数10倍,这也表明玉米生殖类细胞内剪切 后需要RAD51介导的HR进行修复[96,97]。与生殖类细胞剪切修复相比,在体细胞组织转座过程中,转座子剪切后往往会形成多种类型footprint序列,大量及体细胞组织转座事件研究已经证实了这一点[56,93,94]。但体细胞组织内转座子剪切部位绝大多数不存在微同源序列,因此,体细胞组织内转座子剪切后更可能是通过非模板修复方式进行修复的。
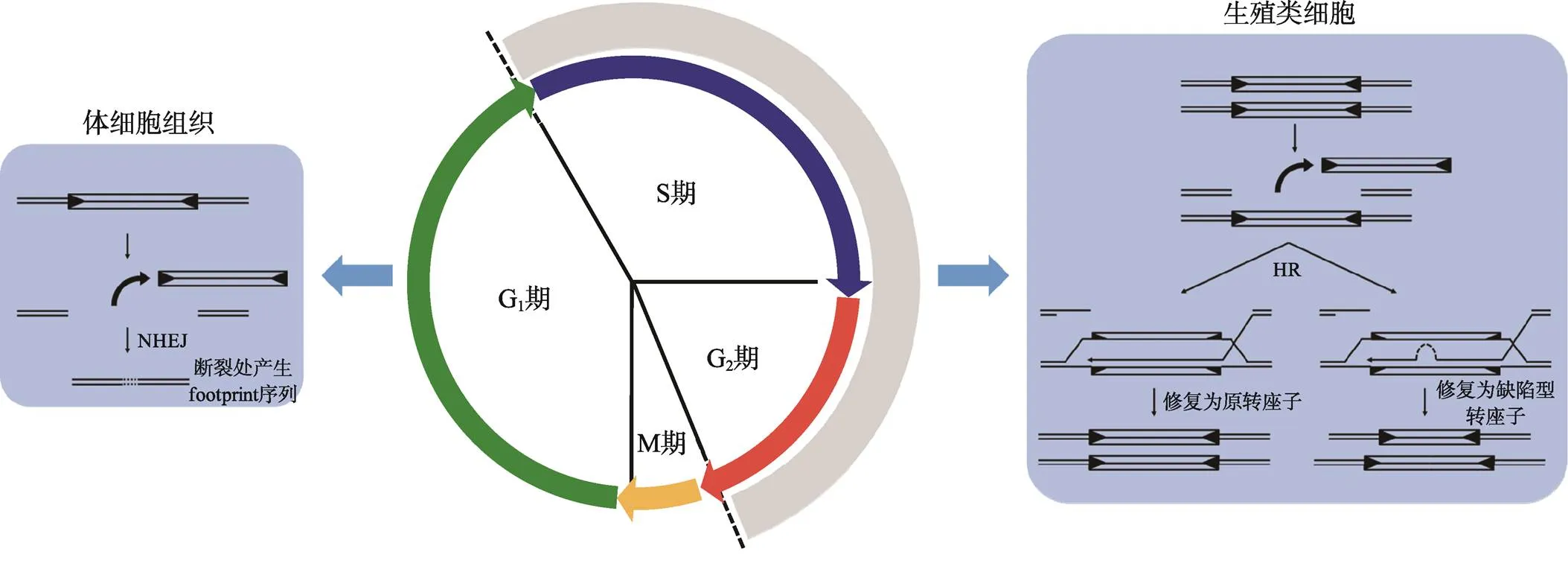
图5 转座子剪切后的DNA双链断裂修复
中间圆形表示细胞周期;左图表示转座子剪切发生在体细胞的S期前,转座子剪切后形成的DSB通过NHEJ方式修复,产生不同类型footprint序列;右图表示转座子剪切发生在生殖类细胞的S期或G2期,转座子剪切后形成的DSB通过HR方式以姐妹染色单体为模板进行修复,在断裂处修复为原转座子或由于微同源序列导致修复链滑移,修复为缺陷型转座子。
3.3 染色体外环形结构
原核转座元件通过闭合环形结构介导转座过程[98],而早期研究发现,在携带活性的玉米材料中,转座子也能够以染色体外共价闭合的环形结构形式存在,并且这些环形的出现依赖于活性的存在,因此,其可能是MURA作用下的转座中间体或是剪切后的产物[99]。但由于环形序列信息的缺乏,难以明确其在转座反应中的作用及生物学意义。Li等[56]对玉米的研究发现,在含有活性的玉米体细胞中可以检测到共价闭合的环形或环形缺陷型结构;序列分析证实,这些环形结构的确是转座子两端共价连接的产物;同时酶切实验表明,所有检测的环形结构并不是两侧TIR末端完美的“头顶头(Head- to-Head)”共价连接,而可能是其他更为复杂的序列组成;除了预期大小的扩增产物,该研究还检测到一些其他扩增产物,克隆测序发现多数产物序列在连接点处缺失转座子单侧或两侧末端序列,缺失长度不等(<100 bp至>2 kb),有些涉及编码转座酶的区段。这些染色体外环形结构可能与转座子某些特性相关,例如转座子剪切后在非连锁位点的再次插入。目前,类似的转座子共价闭合环形结构在其他转座子研究中也有所报道[54,100,101],但不同转座子的染色体外环形结构是否参与转座反应,它们如何发挥作用及其生物学意义仍不明确。
4 插入偏好
超家族转座子在基因组内的转座并非随机插入而具有一定的偏好性。目前通过多个突变系已经获得了数万份玉米插入突变体,这些插入遍布整个玉米基因组[89,102,103],与插入突变体不同,新插入位点与原初插入位点并不连锁。值得注意的是,虽然玉米基因组大部分为反转录转座子序列,但绝大部分插入在基因组低拷贝区域的基因内部或基因附近[103~105]。进一步研究发现,更倾向于插入基因的5′末端,且插入区段序列GC含量较高,这与玉米及其他单子叶植物基因5′末端GC含量略高相对应[106]。另外,插入位点与开放染色质表观遗传标记(如DNA甲基化和组蛋白修饰)紧密相关[107]。近期的研究也表明,真核生物转座子插入位点的选择受染色质结构影响[108]。在玉米W22基因组中,插入位点与两翼序列染色质开放性无明显差异,而插入位点染色质开放性显著增加。此外,和都倾向于插入CG和CHG甲基化程度极低的区域,但插入位点通常与CG和CHG高度甲基化区域相距较远,而插入位点与这些高度甲基化区段距离较近[109],这些特征有助于理解更多插入在基因UTR区而更倾向于插入基因编码区。
5 靶位点重复序列
不同超家族转座子插入基因组后会在其两侧形成一定长度的正向重复序列,这些序列来自插入位点,被称为靶位点重复序列(target site duplication, TSD)。通常同一家族的转座子重新插入后形成相同长度甚至固定组成的TSD,因此,TSD序列长度、固定的序列组成也是进行转座子分类的依据之一[20,21]。超家族转座子转座主要形成长度为9 bp的TSD,并且这些TSD无明显的序列组成规律。近期研究表明,TSD与DNA转座子的转座行为之间关系密切。例如,在异源酵母系统中研究水稻的转座遗传特征时发现,当改变一侧TSD中紧邻的前3个碱基后,转座子的剪切频率显著下降,对于较长的非自主性转座子这种影响更为明显。并且不一致的TSD还会影响非自主性转座子NA剪切位点的精确修复,而对转座子重新插入频率并无显著影响[71]。另外,在异源酵母系统中,TSD同样影响埃及伊蚊的转座行为。当非自主性转座子携带有8 bp或9 bp TSD时,剪切频率较无TSD情况下显著提高,但与不同的是,当携带TSD时,相应转座子重新插入的频率也有所提高。此外,TSD的缺失同样影响相应转座子剪切位点的精确修复。当携带有8 bp或 9 bp TSD时,90%的回复突变均为精确剪切;当无TSD时,精确剪切频率仅占所有回复突变的10%。而对于TSD序列组成的研究表明,不同的TSD序列组成对于转座子的剪切和重新插入均无显著影响[58]。由此可见,TSD序列的有无、一致性及其长度对元件转座行为的影响更大,而TSD的序列组成对于转座行为的影响较小。在植物中超家族转座子的转座反应受到严格调控,而异源酵母系统中开展的研究可能不足以完全涵盖和揭示TSD在植物转座过程中的作用和调控机制。
6 玉米自主性MULEs元件
除了以外,目前玉米中还克隆了另外两个自主性元件:[55]和[56]。这两个转座子与间存在一些共性,例如都具有同源序列,含有较长的TIR,插入位点形成9 bp TSD。而系统进化分析表明,这两个转座子各自作为独立的自主性转座子已经存在了数百万年[73]。与相比,和共享某些特性:(1)玉米中和的拷贝数很低;(2)它们在生殖类细胞内发生回复突变的频率均高于;(3)两者都不含有同源序列[55,56]。更为特殊在生殖类细胞和体细胞组织内剪切后的修复都不留有任何footprint序列。此外,的自主性略显欠缺,目前只检测到玉米e位点的可以发生剪切,并未检测到其重新整合到基因组中,虽然不排除可能与缺少同源基因相关[55],但更可能是两端TIR序列微小差异影响了转座剪切后的再次插入。3′端TIR比5′端TIR在末端少了4个核苷酸(GCTC),生物信息学分析发现,在其他已测序的玉米材料中,-like序列两侧TIR中均含有这4个核苷酸。因此,很可能原本两侧TIR序列一致,在转座到位点过程中3′端TIR发生了序列丢失进而影响到的转座反应,导致其剪切后不能重新插入到基因组中。另外,其他已鉴定的自主性元件同样不含有同源基因,但转座后均可以重新插入到基因组其他位点。因此,即使基因确实与生殖类细胞内转座剪切后的重新插入相关,目前鉴定到的这些元件可能在转座剪切后的重新插入方面进化出了不同的机制,不再需要MURB功能蛋白。玉米中除了以上3个超家族转座子外,遗传学实验还鉴定到另外几个自主性元件,但这些转座子完整的基因组序列目前仍未被克隆,如玉米中家族转座子[110]。已有研究表明,缺陷型()的TIR与TIR的序列在前50 bp高度同源,并且可以使发生移动。但关于自主性转座特性及其与间相互关系目前仍不清楚,尚需进行深入研究。
7 结语与展望
转座子在真核生物基因和基因组的结构及进化过程中扮演着重要角色,众多农作物在其驯化过程中优异农艺形状和优良品质的形成以及对生物胁迫和非生物胁迫的不断适应的遗传基础都与转座子引发的变异密不可分。超家族转座子作为Ⅱ类转座子研究的重要方面,是转座子遗传学及功能基因组学的主要研究对象,而转座子研究中仍有许多科学问题亟待解决,转座子的开发应用更有待加强。因此,继续深入转座子基础遗传学研究并不断开发利用转座子资源必将发挥重要的学术及应用价值。随着高通量测序、生物信息学分析及机器深度学习等新技术的发展,转座子深入研究的成果势必更好地服务和推动生命科学的发展。
[1] Ravindran S. Barbara McClintock and the discovery of jumping genes., 2012, 109(50): 20198–20199.
[2] Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, Imbeault M, Izsvák Z, Levin HL, Macfarlan TS, Mager DL, Feschotte C. Ten things you should know about transposable elements., 2018, 19(1): 199.
[3] Huang CR, Burns KH, Boeke JD. Active transposition in genomes., 2012, 46: 651–675.
[4] Liu Z, Xu JH. The application of the high throughput sequencing technology in the transposable elements., 2015, 37(9): 885–898.刘振, 徐建红. 高通量测序技术在转座子研究中的应用. 遗传, 2015, 37(9): 885–898.
[5] Li SF, Li S, Deng CL, Lu LD, Gao WJ. Role of transposons in origin and evolution of plant XY sex chromosomes., 2015, 37(2): 157–164.李书粉, 李莎, 邓传良, 卢龙斗, 高武军. 转座子在植物XY性染色体起源与演化过程中的作用. 遗传, 2015, 37(2): 157–164.
[6] Li MM, Zhang DF, Gao Q, Luo YF, Zhang H, Ma B, Chen CH, Whibley A, Zhang YE, Cao YH, Li Q, Guo H, Li JH, Song YZ, Zhang Y, Copsey L, Li Y, Li XX, Qi M, Wang JW, Chen Y, Wang D, Zhao JY, Liu GC, Wu B, Yu LL, Xu CY, Li J, Zhao SC, Zhang YJ, Hu SN, Liang CZ, Yin Y, Coen E, Xue YB. Genome structure and evolution ofL., 2019, 5(2): 174–183.
[7] Mascher M, Gundlach H, Himmelbach A, Beier S, Twardziok SO, Wicker T, Radchuk V, Dockter C, Hedley PE, Russell J, Bayer M, Ramsay L, Liu H, Haberer G, Zhang XQ, Zhang Q, Barrero RA, Li L, Taudien S, Groth M, Felder M, Hastie A, ŠimkováH, Staňková H, Vrána J, Chan S, Muñoz-Amatriaín M, Ounit R, Wanamaker S, Bolser D, Colmsee C, Schmutzer T, Aliyeva-Schnorr L, Grasso S, Tanskanen J, Chailyan A, Sampath D, Heavens D, Clissold L, Cao S, Chapman B, Dai F, Han Y, Li H, Li X, Lin C, Mccooke JK, Tan C, Wang P, Wang S, Yin S, Zhou G, Poland JA, Bellgard MI, Borisjuk L, Houben A, Dolezel J, Ayling S, Lonardi S, Kersey P, Langridge P, Muehlbauer GJ, Clark MD, Caccamo M, Schulman AH, Mayer FXK, Platzer M, Close TJ, Scholz U, Hansson M, Zhang G, Braumann I, Spannagl M, Li C, Waugh R, Stein N. A chromosome conformation capture ordered sequence of the barley genome., 2017, 544(7651): 427–433.
[8] Clavijo BJ, Venturini L, Schudoma C, Accinelli GG, Kaithakottil G, Wright J, Borrill P, Kettleborough G, Heavens D, Chapman H, Lipscombe J, Barker T, Lu FH, Mckenzie N, Raats D, Ramirez-Gonzalez RH, Coince A, Peel N, Percival-Alwyn L, Duncan O, Trösch J, Yu G, Bolser DM, Namaati G, Kerhornou A, Spannagl M, Gundlach H, Haberer G, Davey RP, Fosker C, Palma FD, Phillips AL, Millar AH, Kersey PJ, Uauy C, Krasileva KV, Swarbreck D, Bevan MW, Clark MD. An improved assembly and annotation of the allohexaploid wheat genome identifies complete families of agronomic genes and provides genomic evidence for chromosomal translocations., 2017, 27(5): 885–896.
[9] Li FG, Fan GY, Lu CR, Xiao GH, Zou CS, Kohel RJ, Ma ZY, Shang HH, Ma XF, Wu JY, Liang XM, Huang G, Percy RG, Liu K, Yang WH, Chen WB, Du XM, Shi CC, Yuan YL, Ye WW, Liu X, Zhang XY, Liu WQ, Wei HL, Wei SJ, Huang GD, Zhang XL, Zhu SJ, Zhang H, Sun FM, Wang XF, Liang J, Wang JH, He Q, Huang LH, Wang J, Cui JJ, Song GL, Wang KB, Xu X, Yu JZ, Zhu YX, Yu SX. Genome sequence of cultivated upland cotton (TM-1) provides insights into genome evolution., 2015, 33(5): 524–530.
[10] Schmutz J, Cannon SB, Schlueter J, Ma J, Mitros T, Nelson W, Hyten DL, Song Q, Thelen JJ, Cheng J, Xu D, Hellsten U, May GD, Yu Y, Sakurai T, Umezawa T, Bhattacharyya MK, Sandhu D, Valliyodan B, Lindquist E, Peto M, Grant D, Shu S, Goodstein D, Barry K, Futrell-Griggs M, Abernathy B, Du J, Tian Z, Zhu L, Gill N, Joshi T, Libault M, Sethuraman A, Zhang XC, Shinozaki K, Nguyen HT, Wing RA, Cregan P, Specht J, Grimwood J, Rokhsar D, Stacey G, Shoemaker RC, Jackson SA. Genome sequence of the palaeopolyploid soybean., 2010, 463(7278): 178–183.
[11] Schnable PS, Ware D, Fulton RS, Stein JC, Wei F, Pasternak S, Liang C, Zhang J, Fulton L, Graves TA, Minx P, Reily AD, Courtney L, Kruchowski SS, Tomlinson C, Strong C, Delehaunty K, Fronick C, Courtney B, Rock SM, Belter E, Du F, Kim K, Abbott RM, Cotton M, Levy A, Marchetto P, Ochoa K, Jackson SM, Gillam B, Chen W, Yan L, Higginbotham J, Cardenas M, Waligorski J, Applebaum E, Phelps L, Falcone J, Kanchi K, Thane T, Scimone A, Thane N, Henke J, Wang T, Ruppert J, Shah N, Rotter K, Hodges J, Ingenthron E, Cordes M, Kohlberg S, Sgro J, Delgado B, Mead K, Chinwalla A, Leonard S, Crouse K, Collura K, Kudrna D, Currie J, He R, Angelova A, Rajasekar S, Mueller T, Lomeli R, Scara G, Ko A, Delaney K, Wissotski M, Lopez G, Campos D, Braidotti M, Ashley E, Golser W, Kim H, Lee S, Lin J, Dujmic Z, Kim W, Talag J, Zuccolo A, Fan C, Sebastian A, Kramer M, Spiegel L, Nascimento L, Zutavern T, Miller B, Ambroise C, Muller S, Spooner W, Narechania A, Ren L, Wei S, Kumari S, Faga B, Levy MJ, Mcmahan L, Van Buren P, Vaughn MW, Ying K, Yeh CT, Emrich SJ, Jia Y, Kalyanaraman A, Hsia AP, Barbazuk WB, Baucom RS, Brutnell TP, Carpita NC, Chaparro C, Chia JM, Deragon JM, Estill JC, Fu Y, Jeddeloh JA, Han Y, Lee H, Li P, Lisch DR, Liu S, Liu Z, Nagel DH, Mccann MC, Sanmiguel P, Myers AM, Nettleton D, Nguyen J, Penning BW, Ponnala L, Schneider KL, Schwartz DC, Sharma A, Soderlund C, Springer NM, Sun Q, Wang H, Waterman M, Westerman R, Wolfgruber TK, Yang L, Yu Y, Zhang L, Zhou S, Zhu Q, Bennetzen JL, Dawe RK, Jiang J, Jiang N, Presting GG, Wessler SR, Aluru S, Martienssen RA, Clifton SW, Mccombie WR, Wing RA, Wilson RK. The B73 maize genome: complexity, diversity, and dynamics., 2009, 326(5956): 1112–1115.
[12] Paterson AH, Bowers JE, Bruggmann R, Dubchak I, Grimwood J, Gundlach H, Haberer G, Hellsten U, Mitros T, Poliakov A, Schmutz J, Spannagl M, Tang H, Wang X, Wicker T, Bharti AK, Chapman J, Feltus FA, Gowik U, Grigoriev IV, Lyons E, Maher CA, Martis M, Narechania A, Otillar RP, Penning BW, Salamov AA, Wang Y, Zhang L, Carpita NC, Freeling M, Gingle AR, Hash CT, Keller B, Klein P, Kresovich S, Mccann MC, Ming R, Peterson DG, Mehboob-Ur-Rahman, Ware D, Westhoff P, Mayer KF, Messing J, Rokhsar DS. The Sorghum bicolor genome and the diversification of grasses., 2009, 457(7229): 551–556.
[13] Studer A, Zhao Q, Ross-Ibarra J, Doebley J. Identification of a functional transposon insertion in the maize domestication gene., 2011, 43(11): 1160–1163.
[14] Yang Q, Li Z, Li WQ, Ku LX, Wang C, Ye JR, Li K, Yang N, Li YP, Zhong T, Li JS, Chen YH, Yan JB, Yang XH, Xu ML. CACTA-like transposable element inattenuated photoperiod sensitivity and accelerated the postdomestication spread of maize., 2013, 110(42): 16969–16974.
[15] Castelletti S, Tuberosa R, Pindo M, Salvi S. A MITE transposon insertion is associated with differential methylation at the maize flowering time QTL., 2014, 4(5): 805–812.
[16] Wang C, Yang Q, Wang WX, Li YP, Guo YL, Zhang DF, Ma XM, Song W, Zhao JR, Xu ML. A transposon-directed epigenetic change inunderlies quantitative resistance tostalk rot in maize., 2017, 215(4): 1503–1515.
[17] Zhang ZH, Zhang X, Lin ZL, Wang J, Liu HQ, Zhou LN, Zhong SY, Li Y, Zhu C, Lai JS, Li XR, Yu JM, Lin ZW. A large transposon insertion in thepromoter increases stalk strength in maize., 2019, DOI: 10.1105/tpc.19.00486.
[18] Kojima KK. Structural and sequence diversity of eukaryotic transposable elements., 2018, DOI: 10.1266/ggs.18–00024
[19] Finnegan DJ. Eukaryotic transposable elements and genome evolution., 1989, 5(4): 103–107.
[20] Kapitonov VV, Jurka J. A universal classification of eukaryotic transposable elements implemented in Repbase., 2008, 9(5): 411–412, 414.
[21] Wicker T, Sabot F, Hua-Van A, Bennetzen JL, Capy P, Chalhoub B, Flavell A, Leroy P, Morgante M, Panaud O, Paux E, Sanmiguel P, Schulman AH. A unified classification system for eukaryotic transposable elements., 2007, 8(12): 973–982.
[22] Bao WD, Kojima KK, Kohany O. Repbase update, a database of repetitive elements in eukaryotic genomes., 2015, 6: 11.
[23] Xu HE, Zhang HH, Han MJ, Sheng YH, Huang XZ, Xiang ZH, Zhang Z. Computational approaches for identification and classification of transposable elements in eukaryotic genomes., 2012, 34(8): 1009–1019.许红恩, 张化浩, 韩民锦, 沈以红, 黄先智, 向仲怀, 张泽. 真核生物转座子鉴定和分类计算方法. 遗传, 2012, 34(8): 1009–1019.
[24] Hou XG, Zhang X, Guo DL. Identification and analysis methods of plant LTR retrotransposon sequences., 2012, 34(11): 1491–1500.侯小改, 张曦, 郭大龙. 植物LTR类反转录转座子序列分析识别方法. 遗传, 2012, 34(11): 1491–1500.
[25] Shen D, Chen C, Wang SS, Chen W, Gao B, Song CY. Research progress of Tc1/Mariner superfamily., 2017, 39(1): 1–13.沈丹, 陈才, 王赛赛, 陈伟, 高波, 宋成义. Tc1/Mariner转座子超家族的研究进展. 遗传, 2017, 39(1): 1–13.
[26] Robertson DS. Characterization of a mutator system in maize., 1978, 51(1): 21–28.
[27] Robertson DS. Genetic studies on the loss ofmutator activity in maize., 1986, 113(3): 765–773.
[28] Strommer JN, Hake S, Bennetzen J, Taylor WC, Freeling M. Regulatory mutants of the maizegene caused by DNA insertions., 1982, 300(5892): 542–544.
[29] Bennetzen JL. Transposable elementis found in multiple copies only in Robertson's Mutator maize lines., 1984, 2(6): 519–524.
[30] Taylor LP, Walbot V. Isolation and characterization of a 1.7-kb transposable element from a mutator line of maize., 1987, 117(2): 297–307.
[31] Lisch D, Jiang N.andtransposons. In: Bennetzen JL, Hake S, eds. Handbook of Maize: Genetics and Genomics. New York, NY:Springer New York, 2009, 277–306.
[32] Hershberger RJ, Warren CA, Walbot V. Mutator activity in maize correlates with the presence and expression of thetransposable element., 1991, 88(22): 10198–10202.
[33] Chomet P, Lisch D, Hardeman KJ, Chandler VL, Freeling M. Identification of a regulatory transposon that controls thetransposable element system in maize., 1991, 129(1): 261–270.
[34] Qin MM, Robertson DS, Ellingboe AH. Cloning of thetransposable element, a putative regulator of somatic mutability of theallele in maize., 1991, 129(3): 845–854.
[35] Mccarty DR, Settles AM, Suzuki M, Tan BC, Latshaw S, Porch T, Robin K, Baier J, Avigne W, Lai J, Messing J, Koch KE, Hannah LC. Steady-state transposon mutagenesis in inbred maize., 2005, 44(1): 52–61.
[36] May BP, Liu H, Vollbrecht E, Senior L, Rabinowicz PD, Roh D, Pan XK, Stein L, Freeling M, Alexander D, Martienssen R. Maize-targeted mutagenesis: A knockout resource for maize., 2003, 100(20): 11541–11546.
[37] Raizada MN.protocols for maize functional genomics., 2003, 236: 37–58.
[38] Qian YX, Cheng X, Liu Y, Jiang HY, Zhu SW, Cheng BJ. Reactivation of a silenced minimaltransposable element system following low-energy nitrogen ion implantation in maize., 2010, 29(12): 1365–1376.
[39] Lisch D, Chomet P, Freeling M. Genetic characterizationof thesystem in maize: Behavior and regulation oftransposons in a minimal line., 1995, 139(4): 1777–1796.
[40] Lisch D, Freeling M. Loss ofactivity in a minimal line., 1994, 39(4): 289–300.
[41] Slotkin RK, Freeling M, Lisch D.causes the heritable inactivation of thefamily of transposable elements in, 2003, 165(2): 781–797.
[42] Slotkin RK, Freeling M, Lisch D. Heritable transposon silencing initiated by a naturally occurring transposon inverted duplication., 2005, 37(6): 641–644.
[43] Stawujak K, Startek M, Gambin A, Grzebelus D.: A family of-like transposable elements targeting TA microsatellites in., 2015, 143(4): 433–440.
[44] Yan L, Gu YH, Tao X, Lai XJ, Zhang YZ, Tan XM, Wang HY. Scanning of transposable elements and analyzing expression of transposase genes of sweet potato []., 2014, 9(3): e90895.
[45] Gbadegesin MA, Wills MA, Beeching JR. Diversity of LTR-retrotransposons and-like transposons in cassava (Crantz)., 2008, 280(4): 305–317.
[46] van Leeuwen H, Monfort A, Puigdomenech P.-like elements identified in melon,and rice contain ULP1 protease domains., 2007, 277(4): 357–364.
[47] Rossi M, Araujo PG, de Jesus EM, Varani AM, Van Sluys MA. Comparative analysis of-like transposases in sugarcane., 2004, 272(2): 194–203.
[48] Neuvéglise C, Chalvet F, Wincker P, Gaillardin C, Casaregola S.-like element in the yeastdisplays multiple alternative splicings., 2005, 4(3): 615–624.
[49] Chalvet F, Grimaldi C, Kaper F, Langin T, Daboussi MJ., an active-like element in the genome of the fungus., 2003, 20(8): 1362–1375.
[50] Lopes FR, Silva JC, Benchimol M, Costa GG, Pereira GA, Carareto CM. The protistharbors multiple lineages of transcriptionally active-like elements., 2009, 10: 330.
[51] Pritham EJ, Feschotte C, Wessler SR. Unexpected diversity and differential success of DNA transposons in four species ofprotozoans, 2005, 22(9): 1751–1763.
[52] Jacinto DS, Muniz Hdos S, Venancio TM, Wilson RA, Verjovski-Almeida S, Demarco R. Curupira-1 and Curupira-2, two novel-like DNA transposons from the genomes of human parasitesand, 2011, 138(9): 1124–1133.
[53] Marquez CP, Pritham EJ., a new subclass ofDNA transposons found in insect viruses and widely distributed in animals., 2010, 185(4): 1507–1517.
[54] Singer T, Yordan C, Martienssen RA. Robertson'stransposons inare regulated by the chromatin-remodeling gene., 2001, 15(5): 591–602.
[55] Xu NZ, Yan XH, Maurais S, Fu HH, O'Brien DG, Mottinger J, Dooner HK., adistant relative with a paradoxical mobile behavior: excision without reinsertion., 2004, 16(5): 1105–1114.
[56] Li YB, Harris L, Dooner HK., an autonomous and rare maize transposon of thesuperfamily with a high gametophytic excision frequency., 2013, 25(9): 3251–3265.
[57] Gao DY. Identification of an active-like element (MULE) in rice)., 2012, 287(3): 261–271.
[58] Liu K, Wessler SR. Functional characterization of the active-like transposable element,from the mosquito, 2017, 8: 1.
[59] Ming R, Vanburen R, Liu Y, Yang M, Han Y, Li LT, Zhang Q, Kim MJ, Schatz MC, Campbell M, Li J, Bowers JE, Tang H, Lyons E, Ferguson AA, Narzisi G, Nelson DR, Blaby-Haas CE, Gschwend AR, Jiao Y, Der JP, Zeng F, Han J, Min XJ, Hudson KA, Singh R, Grennan AK, Karpowicz SJ, Watling JR, Ito K, Robinson SA, Hudson ME, Yu Q, Mockler TC, Carroll A, Zheng Y, Sunkar R, Jia R, Chen N, Arro J, Wai CM, Wafula E, Spence A, Han Y, Xu L, Zhang J, Peery R, Haus MJ, Xiong W, Walsh JA, Wu J, Wang ML, Zhu YJ, Paull RE, Britt AB, Du C, Downie SR, Schuler MA, Michael TP, Long SP, Ort DR, Schopf JW, Gang DR, Jiang N, Yandell M, Depamphilis CW, Merchant SS, Paterson AH, Buchanan BB, Li S, Shen-Miller J. Genome of the long-living sacred lotus (Gaertn.)., 2013, 14(5): R41.
[60] Ferguson AA, Jiang N.-like elements with multiple long terminal inverted repeats in plants., 2012, 2012: 695827.
[61] Holligan D, Zhang XY, Jiang N, Pritham EJ, Wessler SR. The transposable element landscape of the model legume, 2006, 174(4): 2215–2228.
[62] Hoen DR, Park KC, Elrouby N, Yu Z, Mohabir N, Cowan RK, Bureau TE. Transposon-mediated expansion and diversification of a family of-like genes., 2006, 23(6): 1254–1268.
[63] Jiang N, Bao ZR, Zhang XY, Eddy SR, Wessler SR. Pack-MULE transposable elements mediate gene evolution in plants., 2004, 431(7008): 569–573.
[64] Lisch D.transposons., 2002, 7(11): 498–504.
[65] Yu Z, Wright SI, Bureau TE.-like elements in: structure, diversity and evolution., 2000, 156(4): 2019–2031.
[66] Wang J, Yu Y, Tao F, Zhang JW, Copetti D, Kudrna D, Talag J, Lee S, Wing RA, Fan CZ. DNA methylation changes facilitated evolution of genes derived from-like transposable elements., 2016, 17(1): 92.
[67] Ferguson AA, Zhao D, Jiang N. Selective acquisition and retention of genomic sequences by Pack--like elements based on guanine-cytosine content and the breadth of expression., 2013, 163(3): 1419–1432.
[68] Jiang N, Ferguson AA, Slotkin RK, Lisch D. Pack--like transposable elements (Pack-MULEs) induce directional modification of genes through biased insertion and DNA acquisition., 2011, 108(4): 1537–1542.
[69] Zhao D, Hamilton JP, Vaillancourt B, Zhang W, Eizenga GC, Cui Y, Jiang J, Buell CR, Jiang N. The unique epigenetic features of Pack-MULEs and their impact on chromosomal base composition and expression spectrum., 2018, 46(5): 2380–2397.
[70] Benito MI, Walbot V. Characterization of the maizetransposable element MURA transposase as a DNA-binding protein., 1997, 17(9): 5165–5175.
[71] Zhao D, Ferguson A, Jiang N. Transposition of a rice-like element in the yeast., 2015, 27(1): 132–148.
[72] Raizada MN, Benito MI, Walbot V. Thetransposon terminal inverted repeat contains a complex plant promoter directing distinct somatic and germinal programs., 2001, 25(1): 79–91.
[73] Lisch D.andtransposons., 2015, 3(2): A3–A32.
[74] Tan BC, Chen ZL, Shen Y, Zhang YF, Lai JS, Sun SSM. Identification of an active newtransposable element in maize., 2011, 1(4): 293–302.
[75] Dietrich CR, Cui F, Packila ML, Li J, Ashlock DA, Nikolau BJ, Schnable PS. Maizetransposons are targeted to the 5' untranslated region of thegene and sequences flankingtarget-site duplications exhibit nonrandom nucleotide composition throughout the genome., 2002, 160(2): 697–716.
[76] Bennetzen JL, Springer P, Cresse AD, Hendrickx M. Specificity and regulation of thetransposable element system in maize., 1993, 12(1–2): 57.
[77] Liu K, Wessler SR. Transposition of-like transposable elements (MULEs) resemblesandelements and V(D)J recombination., 2017, 45(11): 6644–6655.
[78] Wang QH, Dooner HK. Remarkable variation in maize genome structure inferred from haplotype diversity at thelocus., 2006, 103(47): 17644–17649.
[79] Eisen JA, Benito MI, Walbot V. Sequence similarity of putative transposases links the maizeautonomous element and a group of bacterial insertion sequences., 1994, 22(13): 2634–2636.
[80] Hua-Van A, Capy P. Analysis of the DDE motif in the Mutator superfamily., 2008, 67(6): 670–681.
[81] Raizada MN, Walbot V. The late developmental pattern oftransposon excision is conferred by a cauliflower mosaic virus 35S-driven MURA cDNA in transgenic maize., 2000, 12(1): 5–21.
[82] Lisch D, Girard L, Donlin M, Freeling M. Functional analysis of deletion derivatives of the maize transposondelineates roles for the MURA and MURB proteins., 1999, 151(1): 331–341.
[83] Aziz RK, Breitbart M, Edwards RA. Transposases are the most abundant, most ubiquitous genes in nature., 2010, 38(13): 4207–4217.
[84] Yuan YW, Wessler SR. The catalytic domain of all eukaryotic cut-and-paste transposase superfamilies., 2011, 108(19): 7884–7889.
[85] Hickman AB, Ewis HE, Li X, Knapp JA, Laver T, Doss AL, Tolun G, Steven AC, Grishaev A, Bax A, Atkinson PW, Craig NL, Dyda F. Structural basis oftransposon end recognition by Hermes, an octameric DNA transposase from., 2014, 158(2): 353–367.
[86] Zhou L, Mitra R, Atkinson PW, Hickman AB, Dyda F, Craig NL. Transposition ofelements links transposable elements and V(D)J recombination., 2004, 432(7020): 995–1001.
[87] Babu MM, Iyer LM, Balaji S, Aravind L. The natural history of the WRKY-GCM1 zinc fingers and the relationship between transcription factors and transposons., 2006, 34(22): 6505–6520.
[88] Mccarty DR, Meeley RB. Transposon resources for forward and reverse genetics in maize. In: Bennetzen JL, Hake S, eds. Handbook of Maize: Genetics and Genomics.New York, NY:Springer New York, 2009, 561–584.
[89] Liang L, Zhou L, Tang Y, Li N, Song T, Shao W, Zhang Z, Cai P, Feng F, Ma Y, Yao D, Feng Y, Ma Z, Zhao H, Song R. A Sequence-Indexed mutator insertional library for maize functional genomics study., 2019, 181(4): 1404–1414.
[90] Hickman AB, Chandler M, Dyda F. Integrating prokaryotes and eukaryotes: DNA transposases in light of structure., 2010, 45(1): 50–69.
[91] Hsia AP, Schnable PS. DNA sequence analyses support the role of interrupted gap repair in the origin of internal deletions of the maize transposon,., 1996, 142(2): 603–618.
[92] Donlin MJ, Lisch D, Freeling M. Tissue-specific accumulation of MURB, a protein encoded by, the autonomous regulator of thetransposable element family., 1995, 7(12): 1989–2000.
[93] Britt AB, Walbot V. Germinal and somatic products ofexcision from thegene of, 1991, 227(2): 267–276.
[94] Doseff A, Martienssen R, Sundaresan V. Somatic excision of thetransposable element of maize., 1991, 19(3): 579–584.
[95] Raizada MN, Nan GL, Walbot V. Somatic and germinal mobility of thetransposon in transgenic maize., 2001, 13(7): 1587–1608.
[96] Li J, Harper LC, Golubovskaya I, Wang CR, Weber D, Meeley RB, Mcelver J, Bowen B, Cande WZ, Schnable PS. Functional analysis of maize RAD51 in meiosis and double-strand break repair., 2007, 176(3): 1469–1482.
[97] Franklin AE, Mcelver J, Sunjevaric I, Rothstein R, Bowen B, Cande WZ. Three-dimensional microscopy of the Rad51 recombination protein during meiotic prophase., 1999, 11(5): 809–824.
[98] Loessner I, Dietrich K, Dittrich D, Hacker J, Ziebuhr W. Transposase-dependent formation of circular ISderivatives inand., 2002, 184(17): 4709–4714.
[99] Sundaresan V, Freeling M. An extrachromosomal form of thetransposons of maize., 1987, 84(14): 4924–4928.
[100] Gorbunova V, Levy AA. Analysis of extrachromosomaltransposable elements., 2000, 155(1): 349–359.
[101] Gorbunova V, Levy AA. Circularizedtransposons: formation, structure and fate., 1997, 145(4): 1161–1169.
[102] Settles AM, Holding DR, Tan BC, Latshaw SP, Liu J, Suzuki M, Li L, O'Brien BA, Fajardo DS, Wroclawska E, Tseung CW, Lai J, Hunter CT, Avigne WT, Baier J, Messing J, Hannah LC, Koch KE, Becraft PW, Larkins BA, Mccarty DR. Sequence-indexed mutations in maize using the UniformMu transposon-tagging population., 2007, 8: 116.
[103] Fernandes J, Dong QF, Schneider B, Morrow DJ, Nan GL, Brendel V, Walbot V. Genome-wide mutagenesis ofL. usingtransposons., 2004, 5(10): R82.
[104] Cresse AD, Hulbert SH, Brown WE, Lucas JR, Bennetzen JL.-related transposable elements of maize preferentially insert into low copy number DNA., 1995, 140(1): 315–324.
[105] Hanley S, Edwards D, Stevenson D, Haines S, Hegarty M, Schuch W, Edwards KJ. Identification of transposon-tagged genes by the random sequencing of-tagged DNA fragments from., 2000, 23(4): 557–566.
[106] Wong GK, Wang J, Tao L, Tan J, Zhang J, Passey DA, Yu J. Compositional gradients ingenes., 2002, 12(6): 851–856.
[107] Liu S, Yeh CT, Ji T, Ying K, Wu H, Tang HM, Fu Y, Nettleton D, Schnable PS.transposon insertion sites and meiotic recombination events co-localize with epigenetic marks for open chromatin across the maize genome., 2009, 5(11): e1000733.
[108] Sultana T, Zamborlini A, Cristofari G, Lesage P. Integration site selection by retroviruses and transposable elements in eukaryotes., 2017, 18(5): 292–308.
[109] Springer NM, Anderson SN, Andorf CM, Ahern KR, Bai F, Barad O, Barbazuk WB, Bass HW, Baruch K, Ben-Zvi G, Buckler ES, Bukowski R, Campbell MS, Cannon E, Chomet P, Dawe RK, Davenport R, Dooner HK, Du LH, Du C, Easterling KA, Gault C, Guan JC, Hunter CT, Jander G, Jiao Y, Koch KE, Kol G, Köllner TG, Kudo T, Li Q, Lu F, Mayfield-Jones D, Mei W, Mccarty DR, Noshay JM, Portwood JN, Ronen G, Settles AM, Shem-Tov D, Shi J, Soifer I, Stein JC, Stitzer MC, Suzuki M, Vera DL, Vollbrecht E, Vrebalov JT, Ware D, Wei S, Wimalanathan K, Woodhouse MR, Xiong W, Brutnell TP. The maize W22 genome provides a foundation for functional genomics and transposon biology., 2018, 50(9): 1282–1288.
[110] Shepherd NS, Rhoades MM, Dempsey E. Genetic and molecular characterization of a--, a new mutable system of., 1989, 10(6): 507–519.
Progress onsuperfamily
Chunsheng Cong1, Yubin Li1,2
Transposable elements (TEs) are fragments of DNA sequence, which can mobile from one locus to another within a genome, often replication in the process. Occupying the main component of the genome, TEs can affect the structure and function of gene and/or genome in a variety of ways, and play an important role in the evolution of the host. Based on the transposition intermediate, eukaryotic TEs can be divided into two classes.Thesuperfamily is found in maize (L.) in the 1970s. As the member of class II elements,superfamily transposons are found in all eukaryote genomes and contain many families with clearly distinguishable genetic characteristics. In addition, these TEs transpose at high rates and preferentially insert in gene-rich and low-repetitive genomic regions leading to the rapid generation of massive novel mutations, therefore, they are in great use of both forward and reverse genetics researches. In this review, we summarize the classification, structure characteristic, transposition mechanism, insertion preference and TSD sequence and other autonomousin maize. Moreover, we discuss the problems faced in TEs’ research and research directions in the future, with a view to discuss possible breakthroughs, future development directions and significant impacts with colleagues in the related research field..
superfamily;family;elements; transposition mechanism; insertion preference; target site duplication
2019-09-27;
2019-12-17
国家自然科学基金面上项目(编号:31871642)资助[Supported by the National Natural Science Foundation of China (No. 31871642)]
从春生,博士研究生,研究方向:生物化学及分子生物学。E-mail: congchunsheng@126.com
李玉斌,研究员,博士生导师,研究方向:生物化学及分子生物学。E-mail: liyubin@caas.cn
10.16288/j.yczz.19-301
2020/1/2 18:26:20
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20191231.1146.001.html
(责任编委: 严建兵)
猜你喜欢
林业科学(2022年1期)2022-03-23 06:56:24
中国蜂业(2021年5期)2021-05-22 02:59:26
浙江农林大学学报(2016年6期)2016-12-12 12:01:32
现代工业经济和信息化(2016年2期)2016-05-17 05:34:16
校园英语·中旬(2016年6期)2016-05-14 19:43:12
湖南工业职业技术学院学报(2016年6期)2016-04-17 06:44:30
新教育时代电子杂志(学生版)(2015年31期)2015-12-20 08:28:53
电子工业专用设备(2015年4期)2015-05-26 09:10:40
汽车维修与保养(2015年8期)2015-04-17 03:33:01
无机化学学报(2014年3期)2014-02-28 17:30:48
