
CML促进高脂诱导糖尿病ApoE-/-小鼠非酒性脂肪肝进展的研究
2020-04-11 03:08
中南医学科学杂志 2020年2期
(江苏大学附属医院 1.心内科,2.病理科,江苏 镇江 212001)
糖尿病是危害人类健康的全球性疾病,预计到2030年患病人数将超过5.5亿[1-2]。研究发现糖尿病与非酒精性脂肪肝(Non-alcoholic fatty liver disease,NAFLD)的发生密切相关。已知在一般人群中,NAFLD患病率在13%~15%之间,而糖尿病NAFLD的患病率则迅猛增加至70%[3-5],研究糖尿病NAFLD发病机制意义重大。AGEs是糖毒性的关键代谢产物,在糖尿病相关并发症中作用显著[6-7];但AGEs在其重要代谢器官肝脏NAFLD发病中的作用尚不明晰。因此,我们拟通过体外、体内实验探讨AGEs关键活性成分Nε-羧甲基赖氨酸(Nε-carboxymethyl-Lysine,CML)在糖尿病性非酒性脂肪肝中的作用,以期为防治非酒精性脂肪提供新思路。
1 材料与方法
1.1 主要材料
HepG2购自中国科学院上海细胞库;CML购自Toronto公司;油酸、棕榈酸购自Sigma公司;高糖DMEM培养基购自Hyclone公司;胎牛血清购自维森特公司;总胆固醇(TC)、甘油三酯(TG)、谷丙转氨酶(ALT)、谷草转氨酶(AST)、超氧化物歧化酶(SOD)、丙二醛(MDA)、谷胱甘肽过氧化物酶(GSH-Px)试剂盒购自南京建成生物公司;兔/鼠通用型Streptavidin-HRP试剂盒购自康为世纪公司。
1.2 细胞培养与分组
HepG2细胞在高糖DMEM培养基含10%FBS和1%青霉素-链霉素溶液,温度为37 ℃、含5%CO2环境下培养,待细胞成片生长至融合状态时,根据实验目前其分组如下:对照组(10%FBS高糖DMEM),FFA组(10%FBS高糖DMEM+0.5 mmol/L FFA),CML低剂量组(10% FBS高糖DMEM+0.5 mmol/L FFA+10 μmol/L CML),CML高剂量组(10% FBS高糖DMEM+0.5 mmol/L FFA+100 μmol/L CML)。干预24h后进行油红O染色以及油红萃取实验。
1.3 油红染色以及油红萃取实验
将HepG2细胞以1×105每孔种于6孔板中诱导24 h后,PBS润洗2遍,用60%异丙醇将每个孔润洗一遍,每孔加入1 mL油红O染色液,染色15 min后,使用60%异丙醇再次润洗一遍,使用苏木素染色1 min,置于显微镜下观察。每孔加入1 mL异丙醇萃取细胞内的油红O染色液,每孔100 μL加入96孔板中,于510 nm处测定其吸光度。
1.4 动物模型建立及分组
6周龄ApoE-/-雄性小鼠购自北京大学实验室。ApoE-/-小鼠予腹腔注射链脲佐菌素40 mg/kg/d连续注射5天。2周后将血糖水平≥300 mg/dL的小鼠纳入本研究。20只6周龄ApoE-/-小鼠随机分为4个实验组,对照组(普食+尾静脉注射生理盐水,n=5),模型组(高脂饮食+尾静脉注射生理盐水,n=5),CML低剂量组(高脂饮食+尾静脉注射CML5mg/kg/day,n=5),CML高剂量组(高脂饮食+尾静脉注射CML 10 mg/kg/day,n=5)喂养8周。实验结束后,小鼠眼球摘除眶后取血,用于检测相关指标。剥离出肝脏组织,多聚甲醛固定肝脏组织并进行组织石蜡切片。
1.5 血清学检测
小鼠进行眶后取血后,室温静置1h后,收集上层血清,使用相关试剂盒检测相关指标包括总胆固醇(TC)、甘油三酯(TG)、谷丙转氨酶(ALT)、谷草转氨酶(AST)、超氧化物歧化酶(SOD)、丙二醛(MDA)、谷胱甘肽过氧化物酶(GSH-Px)按照试剂盒说明书操作。
1.6 肝脏HE染色
采用常规HE染色法,以4%的多聚甲醛固定,制备石蜡包片,连续切片,HE染色,显微镜下观察肝脏脂肪变性。
1.7 免疫组织化学染色
石蜡切片进行常规脱蜡水化、抗原修复后。按照兔/鼠通用型Streptavidin-HRP试剂盒说明书进行CML免疫组化染色,显微镜下观察,并通过Image J进行统计定量。
1.8 统计学分析

2 结 果
2.1 CML促进HepG2细胞内脂质蓄积
通过油红O染色检测细胞内脂质蓄积情况(如图1),与对照组相比,FFA组脂滴数目增加(如图1A、1B),加入不同浓度CML后,显著增加了细胞内脂滴的数量(如图1C、D)。
2.2 肝组织中CML的累积
为进一步验证CML是否累积在肝组织中,我们通过CML免疫组织化学染色,检测肝脏中CML含量(如图2),我们发现相比对照组,HFD组CML染色强度稍增强,随着小鼠尾静脉CML给药浓度的增加,肝脏内CML染色强度逐渐增强。

图1 CML对FFA诱导的HepG2细胞脂质蓄积油红O染色(400×)A:对照组;B:FFA组;C:CML低剂量组;D:CML高剂量组与对照组比较,*P<0.05,与FFA组比较,#P<0.05,与CML低剂量组相比,▲P<0.05(n=3)
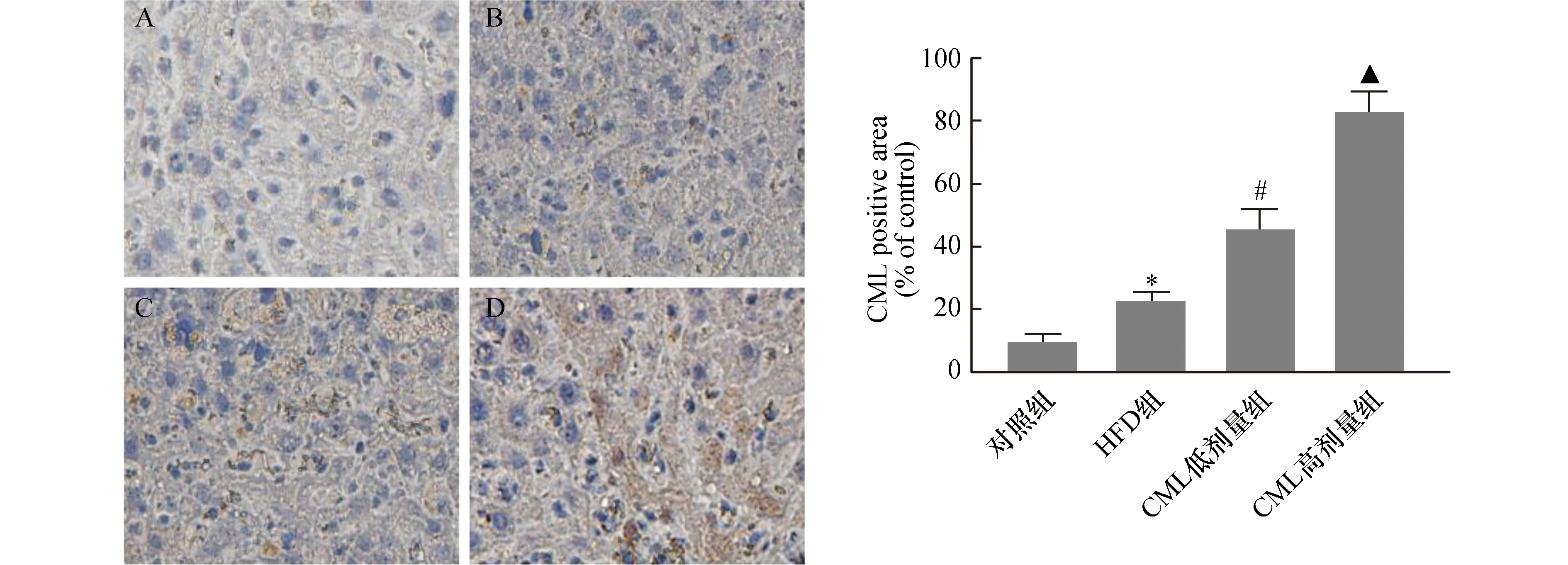
图2 免疫组织化学检测小鼠肝脏CML含量累积图片(400×)A:对照组;B:FFA组;C:CML低剂量组;D:CML高剂量组与对照组比较,*P<0.05,与HFD组比较,#P<0.05,与CML低剂量组相比,▲P<0.05
2.2 CML促进高脂喂养ApoE-/-小鼠脂代谢紊乱
通过细胞实验我们发现CML加重FFA诱导的HepG2细胞内脂质的蓄积。与此同时,我们在体内实验上,我们发现HFD组小鼠血清中TC,TG水平明显升高,CML处理后小鼠血清中TC,TG水平进一步升高。并且这种改变与CML浓度变化相关(如图3)。
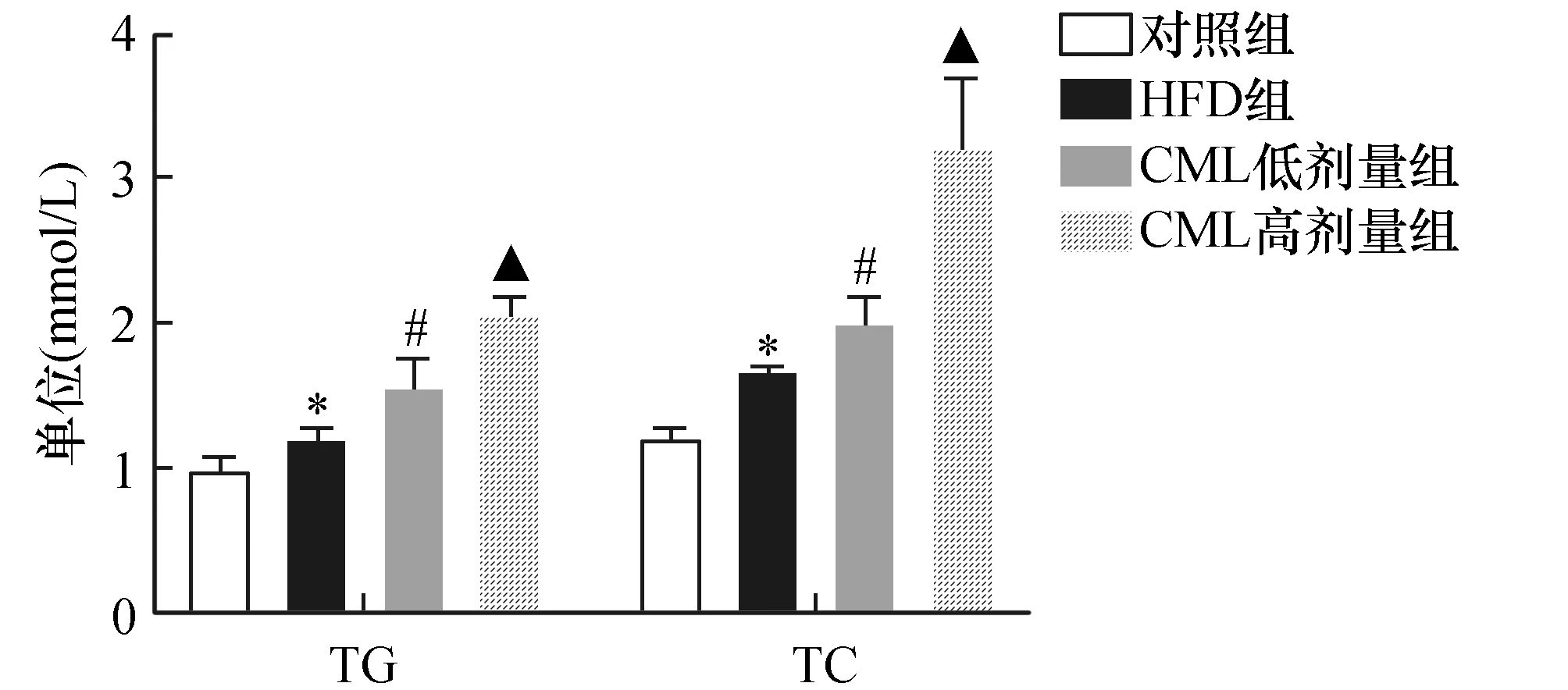
图3 CML促进高脂喂养ApoE-/-小鼠脂代谢紊乱(n=5)ApoE-/-小鼠连续喂养8周,实验结束后ApoE-/-小鼠眶后取血,检测血脂(TC、TG)水平。与对照组比较,*P<0.05,与HFD组比较,#P<0.05,与CML低剂量组相比,▲P<0.05
2.3 CML加重高脂饮食的ApoE-/-小鼠肝脏损伤以及肝脏脂肪样变
与对照组相比,HFD组血清中ALT、ALT活性升高,CML处理后血清中AST、ALT活性明显升高,进一步加重肝脏损伤(图4)肝脏HE染色显示对照组肝叶结构清晰,形态正常,肝细胞成放射状分布,与对照组相比HFD组肝叶紊乱,可见空泡样结构,CML组与HFD组相比肝叶结构辨认不清,肝细胞内可见融合成片的空泡样结构(如图5)。

图4 CML浓度依赖性促进高脂喂养APOE-/-小鼠肝脏损伤(n=5)ApoE-/-小鼠连续喂养8周,实验结束后小鼠眶后取血,检测肝酶(AST、ALT)水平。与对照组比较,*P<0.05,与HFD组比较,#P<0.05,与CML低剂量组相比,▲P<0.05

图5 CML浓度依赖性促进高脂喂养ApoE-/-小鼠肝脏脂肪变性肝脏组织HE染色(200×)观察肝脏病理变化A:对照组;B:HFD组;C:CML低剂量组;D:CML高剂量组
2.4 CML加重高脂喂养ApoE-/-小鼠肝组织内氧化损伤
与对照组比较,HFD组小鼠肝组织中SOD、GSH-Px含量显著减低(P<0.05),MDA含量显著升高(P<0.05);与HFD组比较,CML组肝组织中SOD及GSH-Px含量进一步降低(P<0.05),MDA含量进一步升高(P<0.05)(如图6)。
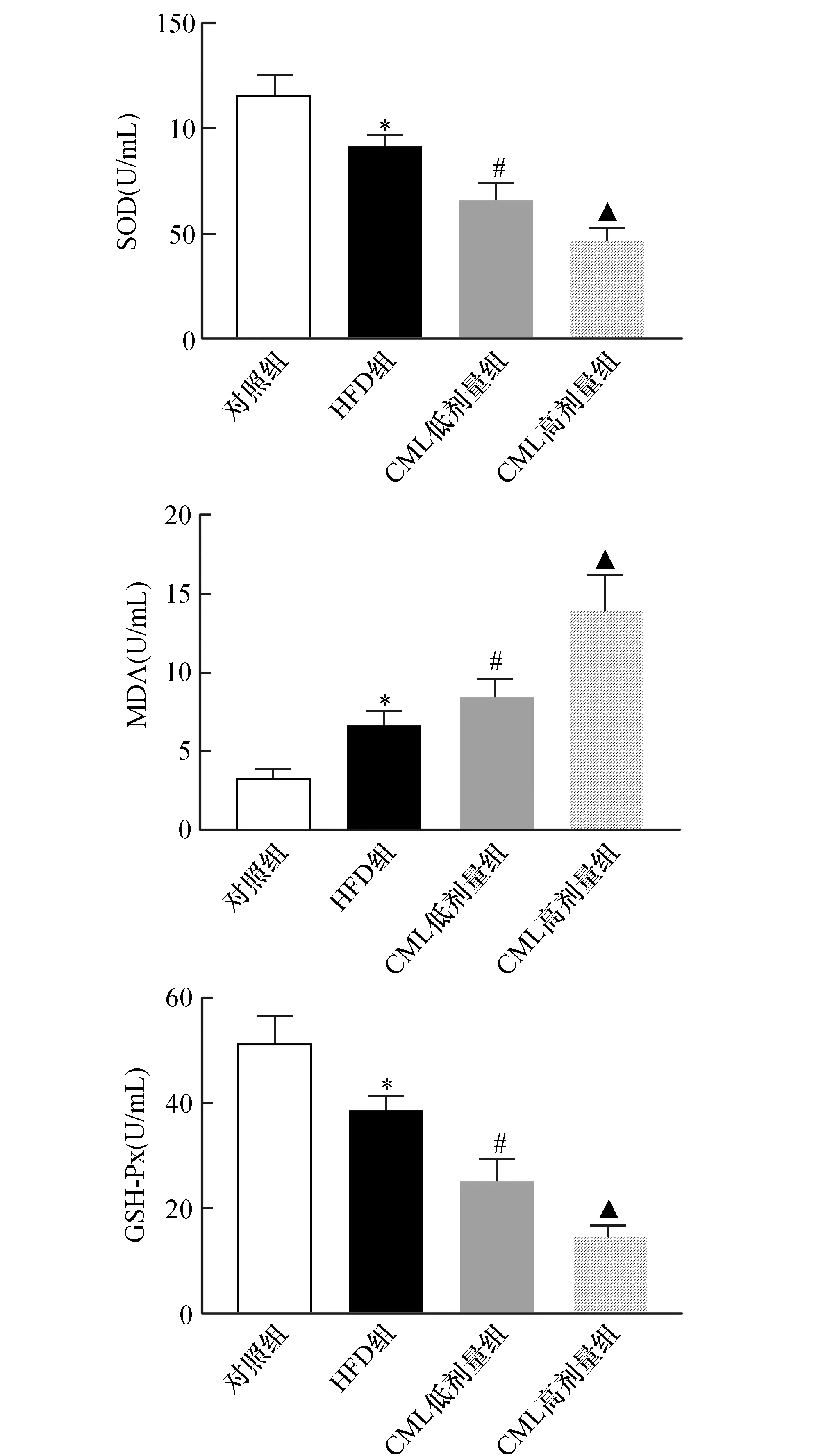
图6 CML浓度依赖性促进高脂喂养ApoE-/-小鼠肝组织内氧化损伤(n=5)ApoE-/-小鼠连续喂养8周,实验结束后小鼠眶后取血,检测SOD、GSH-Px、MDA水平。与对照组比较,*P<0.05,与HFD组比较,#P<0.05,与CML低剂量组相比,▲P<0.05
3 讨 论
糖尿病患者多合并NAFLD发生[8],但由于早期症状体征不明显往往不易察觉,即使患者确诊合并NAFLD的发生,但是部分患者肝酶水平未见明显异常,因此往往造成患者漏诊[9]。糖尿病合并NAFLD发生的患者,其心血管疾病的患病风险明显增加,严重影响患者生活质量。[10-11]
NAFLD是以肝脏细胞空泡性、弥漫性样变为主要病理表现的肝脏疾病。近年来NAFLD的患病率不断上升,据统计在西方国家多达30%的成年人患有NAFLD[12]。而NAFLD的发病机制复杂,目前尚不清楚。“二次打击”是目前学术界普遍接受NAFLD发病机制假说。其中第一次打击主要包括肝脏内脂质的累积,第二次打击主要是在脂质过量累积的基础上肝细胞的氧化损伤。氧化损伤在NAFLD形成中发挥重要作用,因此抑制氧化损伤也是防治NAFLD的重要途径。
为探究CML在糖尿病性非酒性脂肪肝中的作用。我们用HepG2建立体外模型,研究发现CML加重FFA诱导HepG2细胞内脂质的蓄积,并且这种作用依赖于CML浓度变化。为进一步探究CML如何促进NAFLD形成。我们以ApoE-/-小鼠构建体内模型,在体内模型中我们发现CML可明显加重高脂诱导ApoE-/-小鼠血清TC、TG水平,肝脏HE显示CML作用下肝脏组织可见大量空泡样结构。CML刺激下ApoE-/-小鼠血清中AST、ALT水平明显增加。氧化损伤反应是NAFLD重要的发病机制之一,GSH-Px以及SOD可效清除肝组织内的自由基的产生,减少氧化损伤,是肝组织内抑制氧化损伤反应的重要组成部分[13]。而MDA作为一种强毒力的脂质过氧化产物,其水平能反映氧化损伤程度,参与NAFLD的进展[14]。为探究CML是否通过加重氧化损伤促进NAFLD的形成。我们检测了血清中SOD、MDA、GSH-Px水平。结果显示,CML明显上调MDA的表达,抑制SOD、GSH-Px的表达。
我们通过体外、体内实验证明了CML通过促进脂质累积、加重氧化损伤进而促进NAFLD的形成。CML可能作为糖尿病患者NAFLD形成的危险因素,这为糖尿病患者早期预防非酒精性脂肪肝的形成提供了新策略。
猜你喜欢
赣南医学院学报(2022年2期)2023-01-06
基础医学与临床(2022年8期)2022-08-17
中国临床医学影像杂志(2022年6期)2022-07-26
医学食疗与健康(2022年2期)2022-04-23
中国医药科学(2021年18期)2021-01-03
中华养生保健(2020年5期)2020-11-16
计算机应用(2016年10期)2017-05-12
中国新闻周刊(2016年33期)2016-10-27
安徽医科大学学报(2015年9期)2015-12-16
医学研究杂志(2015年12期)2015-06-10
