
碱金属修饰的多孔石墨烯的储氢性能*
2020-04-03 08:44:06元丽华巩纪军王道斌张材荣
物理学报 2020年6期
元丽华 巩纪军 王道斌 张材荣
张梅玲 1)3) 苏俊燕1) 康龙2)
1) (兰州理工大学理学院, 兰州730050)
2) (兰州理工大学材料科学与工程学院, 兰州730050)
3) (兰州大学核科学与技术学院, 兰州730000)
(2019 年 5 月 7 日收到; 2020 年 1 月 15日收到修改稿)
基于第一性原理深入研究了碱金属原子(Li, Na, K)修饰的多孔石墨烯(PG)体系的储氢性能, 并且通过从头算分子动力学模拟了温度对Li-PG吸附的H2分子稳定性的影响. 研究结果表明, PG结构的碳环中心是碱金属原子最稳定的吸附位置, PG单胞最多可以吸附4个碱金属原子, Li原子被束缚最强, 金属原子间无团聚的倾向; H2分子通过极化机制吸附在碱金属修饰的PG结构上, 每个金属原子周围最多可以稳定地吸附3个H2分子; Li-PG对 H2分子的吸附最强 (平均吸附能为–0.246 eV/H2), Na-PG对H2分子的吸附较弱(平均吸附能为–0.129 eV/H2), K-PG 对 H2分子的吸附最弱 (平均吸附能为–0.056 eV/H2), 不适合用做储氢材料; 在不考虑外界压强且温度为300 K的情况下, Li-PG结构可稳定地吸附9个H2分子, 储氢量为9.25 wt.%;在400 K时, 有7个吸附H2分子脱离Li-PG的束缚, 在600—700 K的范围内, 吸附H2分子全部脱离了Li-PG体系的束缚.
1 引言
多孔石墨烯(PG)是一种在石墨烯层状结构中存在纳米孔的二维材料, 纳米孔的大小和几何形状可以被调控[1,2], 不同几何构型的PG展示出金属、半金属及半导体的不同性能. 瑞士科学家Bieri等[3]在2009年首次合成了一种PG结构, 从扫描隧道显微镜图可以清晰地看到纳米孔结构均匀地分配在石墨烯平面内, 或者准确地描述为石墨烯平面内周期性的缺失苯环, 纳米孔间距是 7.4 Å. 这种PG结构的单胞由两个C6H3环组成[4,5], 可以通过3 × 3石墨烯超胞去掉一些碳原子后再利用H原子饱和悬挂键而得到. 利用密度泛函理论[4,5]与晶体轨道法[6]计算分析PG的能带结构表明其属于宽带隙的半导体材料, 这种材料解决了石墨烯0带隙的缺陷. 由于纳米孔洞的存在, PG展现出与纯石墨烯不同的特性, PG在储氢[4]、气体分离或净化[5,7]、DNA测序[8]、超级电容器[9]、水净化[10]等许多领域具有潜在的应用价值.
基于Bieri等[3]实验制备的PG结构, 理论研究表明此结构适合用作储氢材料. Du等[4]利用第一性原理采用VASP软件研究了Li修饰PG的储氢性能, Li原子稳定地吸附在碳环中心, 结合能为1.81 eV, 远高于Li吸附在完整石墨烯的结合能(0.86 eV). Li-PG 结构吸附 12个 H2分子的吸附能为 0.243 eV/H2, 储氢量高达 12 wt.%. Reunchan和Jhi[11]利用 VASP软件, 采用广义梯度近似(GGA)泛函研究了金属修饰PG的储氢性能, 文中只给出了PG和金属原子间的结合能、体系吸附H2分子数目及H2分子吸附能等计算结果, 他们指出Ca-PG最适合做储氢材料, 而Al-PG结构吸附H2较弱, 不适合用作储氢材料. 但Ao等[12]利用Dmol3软件采用局域密度近似(LDA)泛函计算表明Al修饰的PG能够提高H2分子的吸附能力, Al稳定吸附于PG结构的CH环中心上方,Al的结合能为1.78 eV. PG单胞两面最多可吸附两个Al原子, 而且每个Al原子周围最多可以吸附 6个 H2分子, H2的吸附能在 0.41—1.11 eV/H2范围内, 储氢量为 10.5 wt.%. 本课题组[13−15]近年来研究了较重金属原子修饰PG的储氢性能,PG单胞单侧最多可以吸附1个金属原子, 否则金属原子有团聚的倾向.
为了提高金属原子和PG间的结合能力,Lu课题组[16]采用B和N掺杂来改性PG, 利用VASP软件选取GGA泛函研究了Li和Ca修饰PG的储氢性能. 研究结果表明N掺杂使得金属和PG之间的作用变弱, B掺杂虽然可以提高金属和PG之间的作用, 但是掺杂B原子的数目不能超过2个. B掺杂可增强Ca-PG吸附H2分子的强度, 可是B掺杂略减弱Li-PG吸附H2分子的强度(H2分子吸附在Li-B-PG的吸附能为0.08 eV/H2,略低于吸附在 Li-PG上的吸附能0.09 eV/H2).Huang和Miao[17]采用O原子来改性PG (用1个O原子替代PG中的1个H原子), 利用SIESTA软件研究了Li修饰PG体系的储氢性能, Li的结合能高达到 3.84 eV, Li原子周围吸附 5个 H2,H2的吸附能在 0.2—0.5 eV 范围. Wang 等[18]利用N原子改性 PG (PG中连接 H的 C原子和H原子被1个N原子替代, 称之为g-CN), 利用锂和钙原子共同修饰g-CN结构, Li和Ca稳定的吸附位置分别为不完整碳环中心和纳米孔中心, 储氢量为 9.17 wt.%. 最近, Chen 等[19]研究 Li修饰 g-CN层的储氢性能发现单个Li原子和CN层的相互作用很强(结合能为4.73 eV), N掺杂可以提高PG的储氢能力. Wang等[20]对一种PG结构(单胞由C—C键连接3个C6H2环组成, 简称CMP)的储氢性能做了研究, 此结构与 Bieri等[3]的PG结构(单胞由 C—C键连接两个C6H3环组成)很相似, 从头算分子动力学模拟表明在温度为300 K 时, 体系储氢量为 10.89 wt.%. 此外, 进一步的研究[21]表明N掺杂此结构可增强对Li的吸附作用, 体系储氢量得到提高.
PG实验储氢的研究较少, Ning等[22]利用化学气相沉积模板法(template CVD)制备出PG,表面积为 2038 m2·g–1, 纳米孔尺寸约为 1 nm, 在3.1 MPa 和 274 K 的条件下, 可吸附 15.3 mmol/g的H2分子. Xia等[23]研究了不同结构的PG吸附,具有孔表面积大的PG结构吸附能力强, 但储氢量仅有 0.75 wt.% (77 K 和 1 bar (1 bar = 105Pa)),说明纯的PG由于化学惰性使得其与氢气的结合较弱, 需经过金属原子或其他化学官能团修饰才适合 储 氢 . Elyassi等[24]实 验 结 果 表明 , 在 室 温298 K 和压强 34 bar的条件下, PG、石墨烯及氧化石墨烯的储氢量分别是 2.56 wt.%, 1.70 wt.%及0.74 wt.%, 并指出PG的高吸附量是因为其具有高的孔体积、大的比表面积和较小的孔尺寸.
已有研究结果表明Li修饰PG可以增强PG对H2分子吸附能力, 但温度对Li-PG吸附H2分子稳定性的影响以及释放H2的温度没有被进一步讨论, 对于碱金属原子Na和K修饰PG储氢性能的研究很少. 本文主要利用第一性原理系统深入研究了碱金属原子Li, Na, K修饰PG的储氢性能并且利用从头算分子动力学(ab initioMD, AIMD)模拟Li-PG结构吸附的H2分子数目随温度的变化情况.
2 模型与计算方法
本文计算利用CASTEP软件, 考虑自旋极化效应, 选取超软赝势[25], 采用 GGA的 Perdew-Burke-Ernzerhof (PBE)交换关联泛函[26]. 我们对截断能和K点的选择进行了收敛测试, 截断能选取 500 eV,K点为 6 × 6 × 1, 与前人对 PG 相关计算选取的参数一致[4,5]. 几何结构优化无对称性限制, 计算中 SCF 收敛标准选取 1.0 × 10–6eV/atom,能量收敛标准选取 5.0 × 106eV, 结构弛豫止于原子间的力小于0.01 eV/Å. 为了计算中考虑范德瓦耳斯作用, 采用了色散校正DFT-D的Grimme方法(我们在文献[13]中曾讨论了此方法对计算吸附能的影响). 计算模型选取的PG单胞由两个C6H3环构成, 真空层为 18 Å, 利用周期性边界条件.
此外, 我们利用分子动力学研究温度对Li-PG吸附H2分子性能影响时选取了CASTEP中的 Dynamics, 采用 NVT 系综及 Nośe-Hoover恒温扩展方法, 外界压强不考虑, 步长为 1 fs. 因为Dynamics模拟计算非常耗时, Wang[20]和 Ao[27]利用分子动力学计算选取了1.5 ps与1 ps的模拟时长, 为了使计算结果有意义且节约时间, 我们选取的模拟时长为1.5 ps.
碱金属原子和PG间的平均结合能定义为

其中,EnM+PG,EPG与EM对应的是n个碱金属原子修饰PG的总能量、PG单胞总能量以及单个碱金属原子总能量;n为碱金属原子个数.
H2分子的吸附能和平均吸附能分别定义为
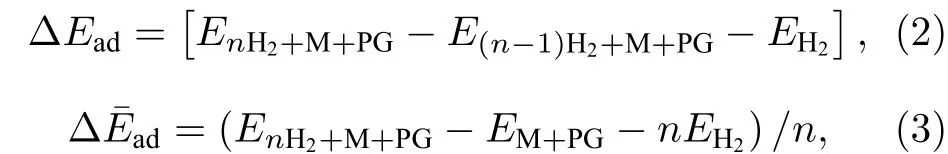
3 结果与讨论
3.1 碱金属原子修饰PG的几何结构和电子结构
首先确定单个碱金属原子吸附在PG上的稳定位置, 图S1 (见补充材料(online))描述了金属原子可能吸附的初始位置. 分别放于不同位置的Li, Na, K 原子, 结构优化后 Li和 Na 占据 h1和 h2位置, K占据 h1和 b2位置, 见表S1 (见补充材料(online)). 通过对碱金属原子的结合能(表S1)做进一步分析, 发现Li原子最稳定的吸附位置是完整 C 环中心 h1, 结合能量值为 1.654 eV (Du[4]采用LDA泛函计算结果为1.81 eV, Rao[28]采用PBE泛函计算结果为 1.38 eV, Lu 等[16]采用 PW91泛函计算结果为1.40 eV); Na位于h1和h2位置的两种几何结构对应的基态能量差别很小(结合能–0.877 eV 和–0.879 eV, 见表S1); K 最稳定的吸附位置是 h1 (结合能–1.037 eV, 见表S1). 图1描绘了碱金属原子吸附在PG上优化后的结构, 图中标出的红色数据是金属原子与碳环中心的距离,Li原子最靠近PG层, 表明Li与PG的相互作用最强. 分析 Mulliken 原子布居, Li带电荷+1.14 e,Na 带 电 荷+0.99 e, K 带 电 荷+1.02 e (Hirshfeld电荷分析, Li带电荷 0.42 e, Na 带电荷 0.35 e,K 带电荷 0.36 e), Li转移到 PG 的电荷最多, 进一步表明Li与PG的相互作用最强. 碱金属原子带正电荷, PG层的C原子带负电荷, 因而PG层和金属原子之间形成静电场, 有利于基体吸附H2分子.

图1 一个碱金属原子修饰 PG 优化后的几何结构(a) Li-PG; (b) Na-PG; (c) K-PGFig. 1. Optimized geometry structure of a alkaline metal atom decorated PG: (a) Li-PG; (b) Na-PG; (c) K-PG.
PG同侧吸附两个金属原子优化后的几何结构如图 S2 (见补充材料 (online))所示, Li原子或K原子位于PG层不同碳环中心, Na原子略微偏离碳环中心. Li-PG与K-PG结构未发生明显的形变, Na-PG 结构发生了微小形变, Li, Na, K 原子的平均结合能分别为–1.670, –1.590, –1.123 eV, 结合能量值均大于内聚能[11,29,30]. 两个吸附Li原子间距及两个吸附K原子间距均为4.329 Å (Du等[4]计算的 Li-Li间距为 4.23 Å), 由 PG 的 2 × 2超胞测得Na原子与其最近邻Na原子的间距为4.057 Å(大于钠晶体最近邻两个Na原子间距3.716 Å),因而PG同侧吸附两个碱金属原子而不会发生团聚现象. PG两侧吸附金属原子优化后的结构如图S3(见补充材料 (online))所示, Li, Na, K 的平均结合能分别是为–1.670, –1.123, –1.161 eV, Li或 K 原子修饰的PG结构未发生明显形变, 但Na-PG几何结构略微形变, 4个Na原子略偏离了完整碳环中心.
3.2 碱金属原子修饰PG吸附H2分子的性能
3.2.1 PG 单侧吸附氢分子的结构
为了明确Li, Na, K修饰对PG吸附H2分子能力的影响, 研究了未掺杂的PG结构吸附H2分子的情况, 1个H2分子被放置于PG的6个不同的初始吸附位置(如图S1所示)分别进行结构优化, 结果发现放于给定初始位置的H2分子不会自动移动到其他位置, PG吸H2分子后结构未发生形变, 第1个H2分子吸附于h1位置(完整碳环中心)能最低 (氢的吸附能为–0.091 eV), H—H 键长保持不变(自由H2的H—H键长为0.753 Å, 此结果和文献[11]计算结果一致); 第2个H2分子稳定的吸附在碳氢环中两H原子之间, 如图S4 (见补充材料(online))所示, H2分子平均吸附能为–0.082 eV/H2; 吸附第 3个 H2分子后平均吸附能为–0.093 eV/H2, 可能是 H2分子间的相互作用使得吸附能量值增大, 于是在PG上继续吸附第4个 H2分子, 平均吸附能为–0.067 eV/H2. 这些H2分子的平均吸附能的量值低于0.1 eV/H2, 说明PG对H2分子的吸附能力很弱, 因而无法保证能有效地储氢.
Li-PG结构单侧吸附H2分子的几何优化结构如图2所示, H2的吸附能、平均吸附能及Li所带电荷如表1所示. 通过分析Mulliken电荷布居, 组成第 1个 H2的两 H 原子带–0.05 e和–0.11 e电荷, Li带+1.23 e电荷, H 原子从 Li得到电荷而带负电, 则H2分子的共价键被极化而变弱, 通过静电场作用被吸附在Li原子周围. H2的吸附能随着吸附H2数目增多而逐渐减小, 第4个H2吸附能低于0.1 eV, 表明Li原子周围可较强地吸附3个H2, 与以前报道一致[4]. 第 4个 H2分子吸附能仅有–0.044 eV, 而 4 个吸附 H2的平均吸附能是–0.225 eV/H2, 如利用平均吸附能判断, Li周围可稳定吸附4个H2. 通过对结果深入分析发现, 第4个 H2的键长 0.754 Å; 通过 Mulliken 电荷布居分析发现, 吸附第4个H2后Li所带的电荷保持不变 (如表1所列), 第 4个 H2的 1个 H原子得到0.01 e的电荷, 另 1个 H 原子没有电荷得失; 通过Hirshfeld电荷分析发现吸附第4个H2前后, Li原子带的电荷保持不变(0.24 e), 而第4个H2的两H原子不带电荷(0 e). 这说明第4个H2属于物理吸附, 不能稳定的吸附在体系上, 因而利用吸附能可以更准确地判断基体最多吸附H2的数目.
位于碳环中心(h1构型)和碳氢环中心(h2构型)的Na原子吸附能相近, 因此考虑这两种结构吸附 H2分子的情况. 图3和图 S5 (见补充材料(online))分别描述了h1和h2构型吸附H2弛豫后的几何结构. 在表2中, DEh1是 h1构型 H2分子的吸附能, DEh2是h2构型H2分子的吸附能.通过分析吸附能, 发现随着H2数目增加, h2构型吸附 H2的能力明显降低. 对于 h1构型, 3个H2可以稳定地吸附在Na周围. 因而, h1构型是吸附H2分子的最佳结构. 分析Mulliken原子布居发现, Na-PG 结构吸附 H2之前, Na 带+0.99 e的电荷, C 均带负电荷. 体系吸附 1个 H2后, Na 带+1.32 e的电荷, H2的两 H 原子带–0.07 e 和–0.15 e的电荷, 表明Na阳离子把0.33 e个电荷转移给H2和C原子. 带负电的C和正电的Na之间形成静电场, H2被极化使得H—H键长由0.753 Å变为0.767 Å.
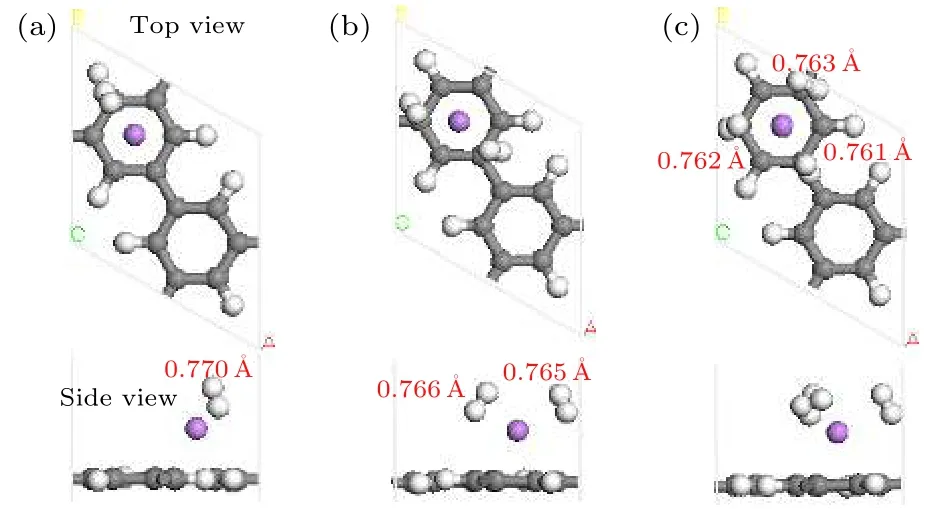
图2 Li-PG吸附氢气分子弛豫后的几何结构(红色字体表示H—H键长)Fig. 2. Optimized geometry structure of the Li-PG with H2 molecules adsorption. Red digits represent the corresponding bond length of H—H.
表1 H2吸附能 (DEad)、平均吸附能 ( )及Li所带的电荷(q)Table 1. Adsorption energies (DEad) and average adsorption energies ( )of H2 molecules, and the charge of Li (q).
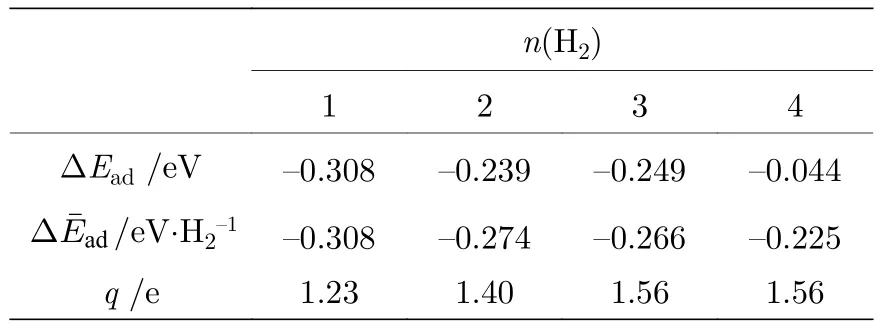
表1 H2吸附能 (DEad)、平均吸附能 ( )及Li所带的电荷(q)Table 1. Adsorption energies (DEad) and average adsorption energies ( )of H2 molecules, and the charge of Li (q).
images/BZ_276_1356_2137_1436_2174.pngn(H2)1234 DEad /eV –0.308–0.239–0.249–0.044 /eV·H2–1–0.308–0.274 –0.266–0.225 q /e1.231.401.561.56
图3 Na-PG 吸附 H2分子优化后的几何结构图 (红色数字表示H—H键长)Fig. 3. Optimized geometry structure of the Na-PG with H2 molecules adsorption. Red digits represent the corresponding bond length of H—H.
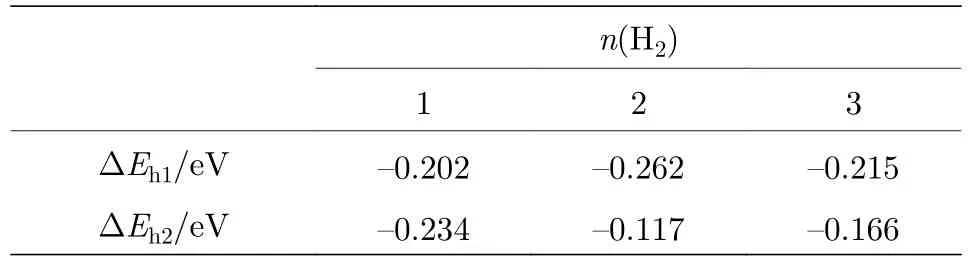
表2 Na-PG 吸附 H2分子的吸附能Table 2. Adsorption energies of H2 molecules on Na-PG.
图4是K-PG吸附1到3个H2优化后的几何结构. 体系吸附的第1个H2垂直于PG平面, 键长为 0.761 Å, 吸附能仅有–0.120 eV 表明 H2被较弱吸附. 当体系吸附第2个H2后, K原子返回碳环中心, 第 2个 H2吸附能为–0.176 eV, 两 H2分子的 H—H键长为 0.763 Å, 均垂直于PG平面.第 3个 H2的吸附能仅有–0.110 eV, 3个 H2垂直于PG平面且位于H原子之上. 随着H2分子数目的增多, H2分子的吸附能逐渐减小, K周围较弱地吸附3个H2分子.
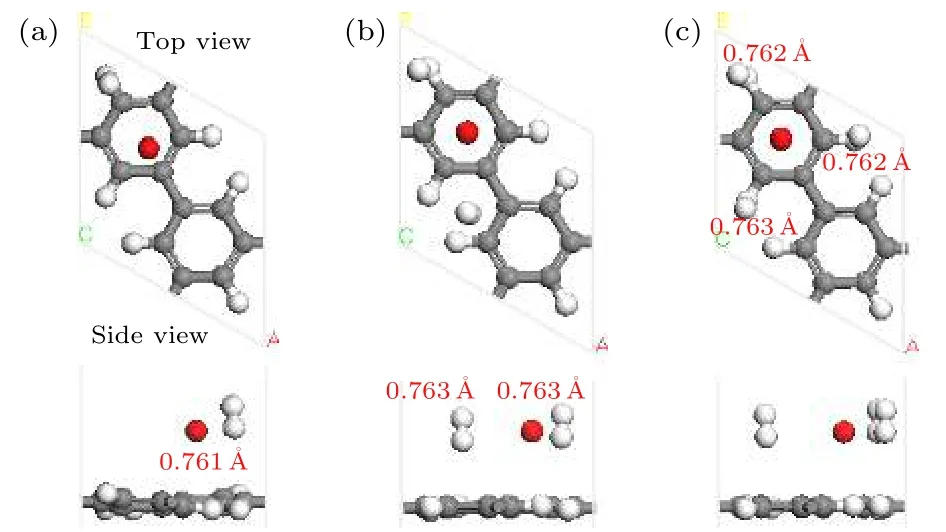
图4 K-PG 结构吸附 H2分子优化后的几何结构图 (红色数字表示H—H键长)Fig. 4. Optimized geometry structure of the K-PG with H2 molecules. Red digits represent the corresponding bond length of H—H.
3.2.2 氢分子的吸附性能
图5和图 S6 (见补充材料 (online))描述了Li-PG, Na-PG, K-PG 吸附 1个 H2的分波态密度图. 氢分子的s轨道和Li原子2s轨道间没有发生轨道耦合现象 (见图5(a)). 从图5(b)看出, H2分子 s轨道出现在费米能级下约–9.1 eV处, Na原子的3s或2p轨道和氢分子的s轨道均没有发生轨道耦合现象. K原子的s和p轨道与H2分子的s轨道也无明显的轨道耦合现象(见图S6). 这说明H2分子吸附在碱金属修饰的PG体系上不是通过Kubas相互作用机制, H2分子主要通过极化机制以及弱范德瓦耳斯相互作用吸附在碱金属修饰的PG上.
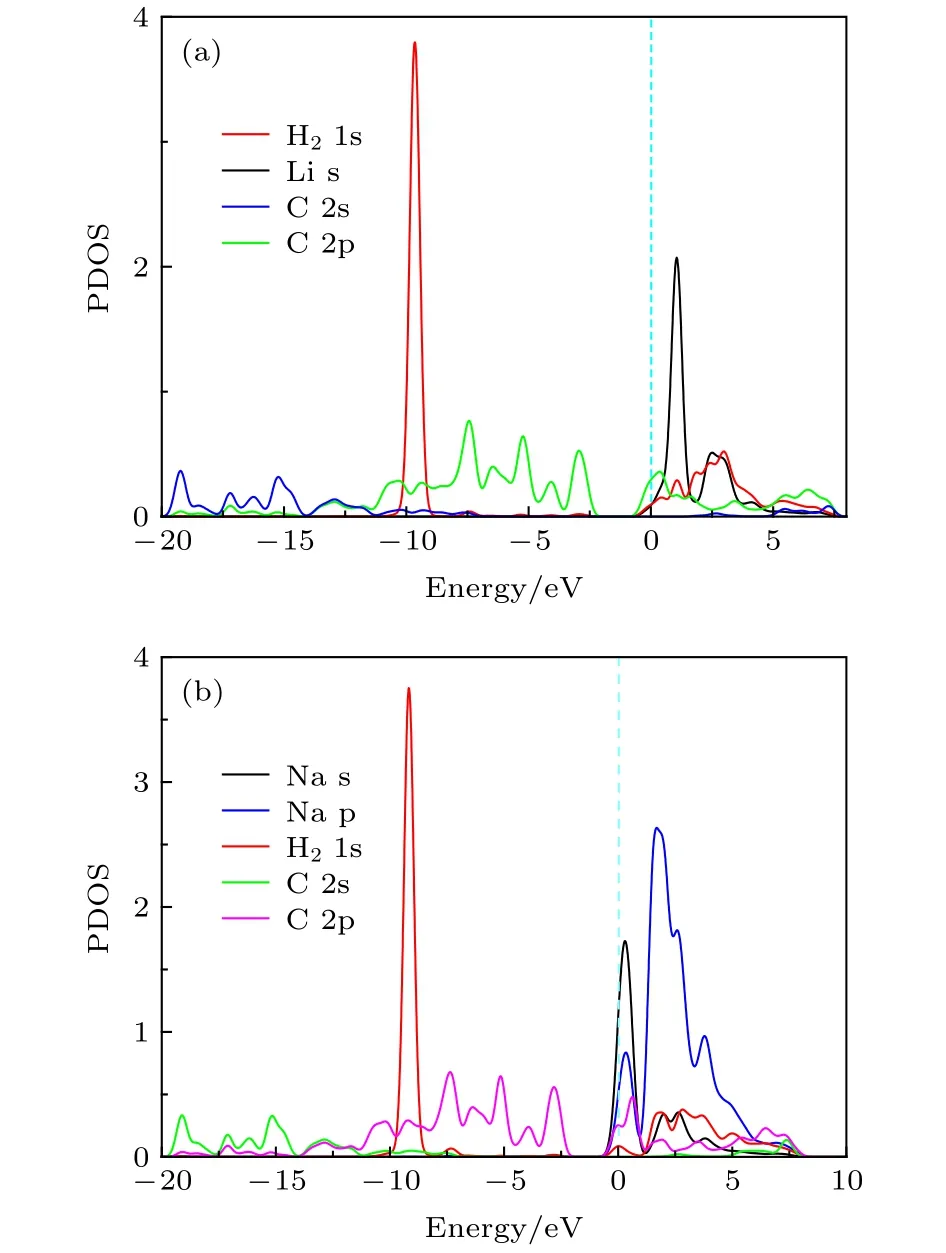
图5 (a) Li-PG 和 (b) Na-PG 结构吸附一个 H2分子的分波态密度图Fig. 5. Partial density of states (PDOS) of a H2 molecule on(a) Li-PG and (b) Na-PG.
为了进一步了解氢的吸附机理, 计算分析了H2吸附体系的差分电荷密度. H2-Li-PG的差分电荷密度如图6及图S7 (见补充材料(online))所示,黄色和蓝色区域代表电荷的减少和聚集. H2分子两侧分别出现了电荷聚集和减少(图6(a)及图S7),表明H2分子被极化, 带正电的Li和带负电的C之间的静电场使氢分子极化, H2分子通过极化机制被吸附到Li-PG体系上[4,18]. 从图6(b)观察到围绕在Li周围的3个H2两边均有电荷聚集和减少, 但远离Li原子的第4个氢分子两侧无电荷分布, 表明Li原子和第4个H2分子间无电荷转移, 这与Mulliken原子布居(见表1)分析一致, 进一步表明Li原子周围可以稳定吸附3个H2分子.以上分析与利用氢分子吸附能判断的结论一致, 说明氢分子的吸附能比平均吸附更能准确地判断吸附 H2分子的数目. Seenithurai等[31]在研究 Li修饰3 × 3石墨烯吸附H2分子时, LDA泛函计算已高估吸附能, 而作者仅通过平均吸附能判断每个Li原子周围可以吸附5个H2分子, 这一结论不准确. 图7描述了Na-PG及 K-PG吸附 1个H2分子差分电荷密度图, 可以看出H2分子通过极化机制被吸附到体系上, 但Na (或K)与 H2分子间聚集的电荷很少, 表明 Na (或K)和H2分子的相互作用较弱.
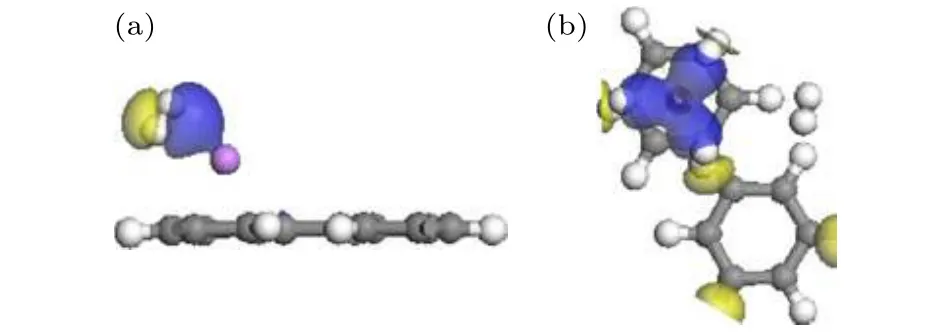
图6 Li-PG吸附H2分子的差分电荷密度图(电荷密度等值面是 0.01 e / Å3)(a) n = 1; (b) n = 4Fig. 6. Charge density differences of n H2 adsorbed on Li-PG system for (a) n = 1 and (b) n = 4. The isovalue is taken to be 0.01 e/Å3.
3.2.3 碱金属修饰的 PG单胞两侧吸附氢分子的情况
PG单胞两侧最多可以吸附4个碱金属原子,最多可以吸附12个H2分子. Li-PG吸附H2分子优化后的稳定几何结构如图8所示, 在该结构中12个H2分子都倾斜的分布于Li的周围, 平均吸附 能 为 –0.246 eV/H2(Du 等[4]的 计 算 结 果 为–0.243 eV/H2), 储氢量为 12 wt.%. Na-PG 吸附H2分子优化后的稳定几何结构如图S9 (见补充材料(online))所示, H2分子的平均吸附能为–0.129 eV/H2, 表明 Na-PG 体系对 H2分子的吸附弱于Li-PG体系. K-PG吸附H2分子优化后的几何结构如图 S10 (见补充材料 (online))所示, 氢分子的平均吸附能为–0.056 eV/H2. 尽管 4个 K 原子可以稳定地被吸附在PG单胞上, 但H2分子被K-PG结构吸附较弱, 室温下不能稳定被吸附, 因而K-PG不适合用作储氢材料.
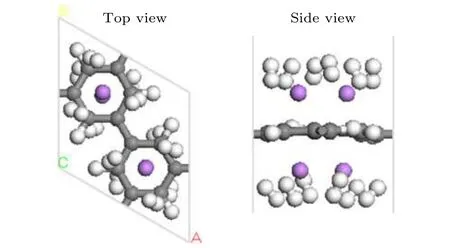
图8 Li-PG结构吸附12个H2分子优化后的几何结构图Fig. 8. Optimized geometry structure of the Li-decorated PG with 12 H2 adsorption.
以上计算忽略了零点能(ZPE), 为了考察ZPE对能量的贡献, 利用CASTEP软件通过计算声子得到ZPE (针对最适合储氢的Li-PG结构进行了计算). 采用不同的方法计算了PG的ZPE,其中线性响应(linear response)法计算的ZPE为3.674 eV, 有限位移 (finite displacement)法计算的ZPE为3.695 eV, 这两种方法计算的ZPE值相差很小, 由于之前的计算采用超软赝势, 所以采用有限位移法. Li-PG 结构的 ZPE 为 3.730 eV, 而单个Li原子的ZPE为0 eV, 所以ZPE对Li的结合能的贡献仅有 0.035 eV (即: 如考虑 ZPE 校正,1个 Li原子与 PG 间的结合能为–1.619 eV). 利用有限位移法计算的 H2分子的ZPE为 0.289 eV(Sacchi和Jenkins[32]计算得到的H2分子ZPE为0.273 eV), Li-PG 吸附 1个 H2分子后的 ZPE 为4.127 eV, ZPE对第 1个 H2分子吸附能的贡献为0.108 eV (即: 考虑 ZPE 校正, 吸附在 Li-PG 上的第 1个 H2分子的吸附能为–0.2 eV). 计算结果说明计算H2分子的吸附能需要考虑ZPE校正.
为了解最适合储氢的Li-PG结构在室温下吸附 H2分子的能力, 通过 AIMD模拟12 H2-Li-PG在不考虑压强且温度300 K下的稳定性. 图9(a)描述了经过AIMD模拟后12 H2-Li-PG平衡结构,Li-PG结构未发生明显形变, 其中的 3个 H2(黄色)脱离体系束缚, 这3个H2和最近邻Li的距离均超过 4 Å. 因此, 在室温下, Li修饰的 PG 单胞最多可吸附 9个 H2, 储氢量为 9.25 wt.%. 总能量随模拟时间的函数关系如图9(b)所示, 图中的曲线在很小能量范围内发生振动, 没有发生大幅度涨落, 进一步表明在室温下Li-PG结构是稳定的. 对12 H2-Na-PG 做了 AIMD 模拟, 在温度为 300 K时, 有5个H2分子脱体系的束缚(图S11 (见补充材料 (online))), 储氢量为 5.47 wt.%.

图9 经过 AIMD 模拟后 12 H2-Li-PG 平衡结构 (a)及总能随模拟时间变化的函数关系(b)Fig. 9. Equilibrated structure of the 12 H2-Li-PG (a) and fluctuations of total energy as a function of simulation time(b) in AIMD simulations at 300 K.
此外, 我们通过AIMD模拟研究了不同温度对吸附H2分子稳定性的影响. 在温度较低时(100 K 和 150 K), H2分子仍较强的吸附在 Li原子周围, 随着温度的升高, 1个H2分子开始远离Li原子, 在 250 K 时, 有两个 H2分子脱离 Li原子的束缚, 如图 S12 (见补充材料 (online)) 所示. 温度继续升高, 更多的H2分子脱离体系的束缚, 在400 K 时, 有 7 个 H2分子脱离体系的束缚, 即58%的H2分子被释放; 随着温度的升高, 释放的H2分子增多, 在600—700 K范围内所有的H2分子全部被释放.
4 结论
本文深入研究了Li, Na, K分别修饰PG单胞的储氢性能. PG单胞最多可吸附4个碱金属原子,借助H2分子吸附能、分波态密度、Mulliken原子布居及差分电荷密度计算分析, 我们发现最多有3个H2分子可稳定吸附在碱金属原子周围, H2分子被吸附是利用极化机制, 氢的吸附能比平均吸附能可以更能准确地判断体系吸附的H2分子数目.Li对H2分子的吸附最强, K对H2分子的吸附最弱, 因而K-PG不适合储氢. Li-PG体系在理想条件下的储氢量高达12 wt.%, 为了进一步明确温度对Li-PG体系吸附H2分子的性能的影响, 通过AIDM模拟计算了12 H2-Li-PG结构随温度变化的情况. 在温度为300 K且不考虑外界压强, 最多有9个H2分子被吸附在Li修饰的PG单胞上, 储氢量为 9.25 wt.%. 随着温度的升高, H2分子相继脱离了Li原子的束缚, 在400 K时, 有58%的H2分子被释放, 在600—700 K范围内所有的H2分子全部被释放.
猜你喜欢
大学物理(2022年9期)2022-09-28 01:10:52
计算机应用与软件(2022年2期)2022-02-19 10:24:04
计算力学学报(2021年3期)2021-07-01 06:07:36
物理通报(2020年7期)2020-07-01 09:28:02
纤维复合材料(2018年2期)2018-12-07 00:41:18
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
文理导航(2015年26期)2015-09-29 14:12:24
中学化学(2015年5期)2015-07-13 07:41:41
中学化学(2015年5期)2015-07-13 07:36:59
湖南师范大学自然科学学报(2015年1期)2015-02-27 14:50:04
