
解淀粉芽孢杆菌mtnN基因对其生物合成 S-腺苷甲硫氨酸的影响
2020-04-02 03:32阮丽英温志友魏雪团
食品科学 2020年6期
阮丽英,温志友,魏雪团,
(1.华中农业大学食品科学技术学院,湖北 武汉 430070;2.爱荷华州立大学食品科学与人类营养系,美国 爱荷华州 埃姆斯 50013)
S-腺苷甲硫氨酸(S-adenosylmethionine,SAM)是广泛存在于生物体内的一种重要的生物活性物质,参与转甲基、转硫和转氨丙基等多种类型的生化反应[1-2],从而影响体内核酸、蛋白质和磷脂等物质的合成与代谢[3-4]。 SAM显著影响细胞代谢活动,充足的SAM是维持机体正常运转的重要保证,缺乏SAM会出现一系列不良症状[5-6]。 研究表明,SAM对肝病、关节炎及抑郁症等疾病具有良好的预防和治疗效果[7-11]。因此,开发SAM相关的功能食品和药品具有重要的价值。
SAM可通过微生物发酵生产,由微生物体内的S-腺苷甲硫氨酸合成酶催化底物L-甲硫氨酸和ATP合成[12]。 目前S A M 的研究主要集中在酵母菌、大肠杆菌(Escherichia coli)和谷氨酸棒杆菌,多通过强化微生物表达S-腺苷甲硫氨酸合成酶,进而将更多的L-甲硫氨酸转化为SAM[13-14]。如He Junyun等[15]在毕赤酵母中强化表达外源S-腺苷甲硫氨酸合成酶基因SAM2,并敲除SAM分支途径中的β-胱硫醚合成酶基因,经发酵条件优化后,工程菌在5 L发酵罐中SAM产量高达13.5 g/L。然而,以甲硫氨酸为底物成本较高,且甲硫氨酸转化率较低(15%~42%),导致SAM价格高[16-17]。有研究者通过葡萄糖或天冬氨酸为原料合成SAM。如Chen Yawei等[18]以葡萄糖为底物从头合成SAM,通过调控ATP和NADPH强化了E. coli合成SAM的能力,然而目前的产量不足10 mg/L; Han Guoqiang等[19]敲除了谷氨酸棒杆菌SAM支链途径和抑制因子基因(mcbR、thrB和Ncgl2640),强化表达了血红蛋白基因和甲硫氨酸合成酶基因,以葡萄糖为底物,SAM产量达到196.7 mg/L。总体而言发酵产量较低,有待进一步提高。
解淀粉芽孢杆菌(Bacillus amyloliquefaciens)具有食用安全、生长快速、易于培养等优点,目前已广泛应用于食品、医药、化妆品和农业领域[20-21]。近年来,随着芽孢杆菌系统生物学和基因操作工具的发展,极大推动了芽孢杆菌代谢工程的发展[22-23]。KEGG数据库显示,B. amyloliquefaciens中存在完整的SAM代谢途径,然而还鲜见通过B. amyloliquefaciens发酵生产SAM的报道。在B. amyloliquefaciens中,基因mtnN编码S-腺苷高半胱氨酸核苷酶(S-adenosylhomocysteine nucleosidase,SAHN,EC 3.2.2.9),如图1所示,该酶既可以参加SAM循环途径,又可以参加甲硫氨酸循环途径,这两个循环途径都消耗SAM[24]。因此,提出假设:阻断或降低mtnN基因的表达可同时抑制SAM循环途径和甲硫氨酸循环途径,从而促进SAM的积累。为验证这个假设,本研究通过基因敲除技术和反义RNA抑制技术构建系列工程菌株,考察mtnN基因对B. amyloliquefaciens合成SAM的影响,阐释了mtnN基因对SAM合成的影响规律,为SAM高产菌株的代谢工程育种提供了新的思路。
1 材料与方法
1.1 材料与试剂
1.1.1 菌株与质粒
本研究所用到的菌株和质粒如表1、2所示。其中 E. coli DH5α用于构建载体。
1.1.2 聚合酶链式反应(polymerase chain reaction,PCR)引物
本研究所用的主要引物见表3,引物合成和质粒测序由武汉擎科创新生物科技有限公司和北京奥科鼎盛生物科技有限公司完成。
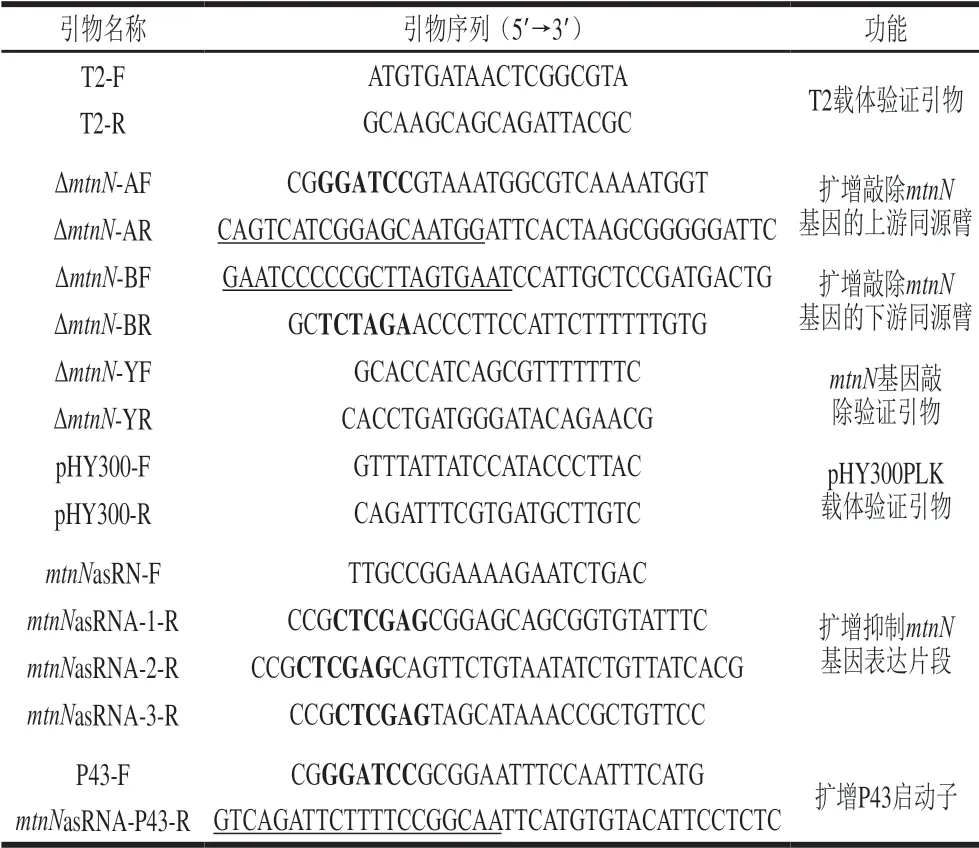
表 3 实验所用PCR引物Table 3 Sequences of primers used for PCR in this study
1.1.3 培养基
Luria-Bertani(LB)培养基:蛋白胨10.0 g/L,酵母浸出粉5.0 g/L,氯化钠10.0 g/L,pH 7.2~7.4,固体培养基加琼脂1.5%。
B. amyloliquefaciens电转化感受态制备所用生长培养基:LB培养基中添加0.5 mol/L山梨醇;洗涤培养基:0.5 mol/L甘露醇,0.5 mol/L山梨醇,10%(体积分数)甘油;恢复培养基:LB培养基中含有0.5 mol/L山梨醇,0.38 mol/L甘露醇。
SAM发酵培养基:蔗糖80 g/L,蛋白胨10 g/L,玉米浆5 g/L,天冬氨酸3 g/L,尿素2 g/L,(NH4)2SO46.3 g/L, NaCl 2.5 g/L,KH2PO43 g/L,MgSO4·7H2O 4.2 g/L,pH 6.5~6.7。
1.1.4 工具酶和试剂
Trans Start®FastPfu DNA聚合酶、Trans Start®easy Taq DNA聚合酶 北京全式金生物技术有限公司;dNTPS、DNA限制性内切酶、T4 DNA连接酶、DL5000 Marker 日本TaKaRa公司;琼脂糖 西班牙Spanish公司;DNA抽提试剂盒 武汉楚诚正茂科技工程有限公司;DNA回收试剂盒和质粒抽提试剂盒 美国Omega BioTek公司;卡那霉素(kanamycin,Kan)、四环素(tetracycline,Tet)、溶菌酶、山梨醇、甘露醇(Biosharp进口分装) 武汉楚诚正茂科技工程有限公司; 甲醇为色谱级,其他均为国产分析级纯或进口分装。
1.2 仪器与设备
1260高效液相色谱仪 美国Agilent公司;AL204电子天平 梅特勒-托利多仪器(上海)有限公司;Alpha Imager EP凝胶成像系统 美国Alpha Innotech公司;AR5120通用型精密天平 美国奥豪斯公司;CR21G高速冷冻离心机 日本Hitachi公司;DYY-8C型电泳仪 北京六一仪器厂;H Q L 3 0 0 B 恒温大幅振荡摇床 武汉中科科仪技术发展有限责任公司;Legend Micro17R高速冷冻离心机 美国Thermo Fisher Scientific公司; MLS-3750高压灭菌锅 日本Sanyo公司;QL-861涡旋振荡器 海门市其林贝尔仪器制造有限公司;Research®固定量程移液器 德国艾本德股份公司;SKP-02.420电热恒温培养箱 黄石市恒丰医疗器械有限公司;SW-CJ-2F 洁净工作台 苏净集团苏州安泰空气技术有限公司;Thermal Cycler (My Cycler) PCR仪 美国Bio-Rad公司;722型分光光度计 上海奥谱勒仪器有限公司;DELTA 320 pH计 梅特勒-托利多仪器(上海)有限公司。
1.3 方法
1.3.1 分子水平操作
E. coli质粒的小量抽提:美国Omega BioTek公司的质粒提取试剂盒,参照说明书中提取步骤。
PCR扩增:所用引物见表3,依次加入常规PCR体系(表4)各成分,混匀后进行扩增反应。反应程序:95 ℃预变性5 min;95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸1 min,30 个循环;72 ℃延伸5 min;12 ℃保持5 min。其中,退火温度依据引物的Tm值,延伸时间依据基因片段大小和聚合酶扩增效率。

表 4 常规PCR体系Table 4 Reaction system of conventional PCR
SOE-PCR依次加入各成分(表5),混匀后进行扩增反应。SOE-PCR程序:95 ℃预变性5 min,95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸1 min,先运行8 个循环,后运行:95 ℃变性30 s,55 ℃退火30 s,72 ℃延伸1 min,30 个循环,72 ℃延伸5 min,12 ℃保持5 min。其中,退火温度依据引物的Tm值,延伸时间依据基因片段大小。

表 5 SOE-PCR体系Table 5 Reaction system of SOE-PCR
DNA的纯化及回收:按照纯化及胶回收试剂盒(美国Omega BioTek公司)说明书上的步骤纯化回收DNA。
1.3.2 mtnN基因缺失菌株的构建
重组载体的构建:选取m t n N 基因上下游序列约500 bp为同源臂片段,设计上游同源臂片段引物(ΔmtnN-AF和ΔmtnN-AR)和下游同源臂片段引物(ΔmtnN-BF和ΔmtnN-BR)。以B. amyloliquefaciensHZ-12的基因组DNA作为模板,分别扩增得到上下游同源臂片段,产物纯化回收后作为两段同源臂SOE-PCR的模板,用引物ΔmtnN-AF和ΔmtnN-BR进行扩增,产物纯化回收后与敲除载体T2(2)-ori同时用BamHI和XbaI进行双酶切,产物回收后用T4连接酶4 ℃连接过夜,连接产物转化至E. coli DH5α感受态细胞,挑取单菌落进行菌落PCR验证,确定为阳性克隆后,提取质粒测序,构建成功的敲除载体为T2(2)-oriΔmtnN。
感受态的制备:B. amyloliquefaciensHZ-12平板划线,挑取适量菌体接种于5 mL液体LB培养基,37 ℃、180 r/min培养8 h;培养物转接于50 mL生长培养基中,37 ℃、250 r/min培养3 h,使OD600nm达1.5~2.5;将培养液倒入无菌的50 mL离心管中,置冰上预冷10 min,6 500 r/min离心5 min收集菌体;用20 mL预冷的洗涤培养基洗涤菌体3~4 次,6 500 r/min离心5 min,弃上清液,将菌体重悬于800 μL洗涤培养基中,每100 μL分装于1.5 mL离心管中,-80 ℃保存备用。
电转化:将5~10 μL质粒DNA(50 ng/μL)加入B. amyloliquefaciensHZ-12感受态细胞,轻柔混匀后转移到预冷的2 mm电转化杯中,冰浴10 min后,电脉冲转化仪2.4 kV电击一次,迅速加入800 μL恢复培养基,于37 ℃、100 r/min恢复培养3 h,涂布于含Kan抗生素平板,37 ℃静置培养16~18 h。挑取转化子单菌落划线于Kan抗性平板,培养10~12 h后进行菌落PCR验证。
单交换菌株的筛选及验证:验证正确的阳性转化子接种于含Kan抗生素的LB液体培养基中,45 ℃、 180 r/min培养12 h;培养物稀释10-6倍涂布Kan抗性平板,45 ℃恒温培养箱中静置培养;挑单菌落划线于Kan抗性平板,45 ℃培养10 h左右,用验证引物和T2引物进行菌落PCR验证。
双交换菌株的筛选及验证:获得的单交换菌株在5 mL LB液体培养基中连续传代,每代12 h,每次取100 μL前一代培养物接种于5 mL LB液体培养基中,37 ℃振荡培养;将最后一代培养物稀释105~106倍,涂布LB平板;对其上长出的单菌落分别对应点种于LB平板和含Kan的LB平板上;选取在LB平板生长而在含Kan的平板不生长的单菌落进行PCR验证,筛选双交换菌株。
1.3.3 反义RNA抑制mtnN基因表达工程菌的构建
重组载体的构建:通过引物m t n N a s R N A-F 和mtnNasRNA-1-R,以互补于目标基因的5’端不翻译序列和目标基因编码区的序列为模板进行扩增,通过NcoI和XhoI对具有反义RNA序列的pUC19-asRNA质粒和PCR得到的目的片段asRNA分别进行双酶切,然后将酶切后的质粒和片段连接进行转化,得到含有目的片段asRNA的质粒pUC19-mtnNasRNA。通过引物P43-F和mtnNasRNAP43-R扩增P43启动子,通过引物mtnNasRNA-F和rrnB-R(质粒本身含有的终止子下游引物),以质粒pUC19-mtnNasRNA为模板进行扩增,PCR产物回收纯化后,以引物P43-F和rrnB-R进行SOE-PCR,用BamHI和XbaI对得到的片段进行双酶切,回收纯化后与同样经过双酶切纯化后的穿梭质粒pHY300进行酶连,转化后得到质粒pHYmtnNasRNA-1。通过引物P43-F和mtnNasRNA-P43-R扩增P43启动子,通过引物mtnNasRNA-F和mtnNasRNA-2-R(mtnNasRNA-3-R),以质粒pHY-mtnNasRNA-1为模板进行扩增,产物回收纯化后与pHY-mtnNasRNA-1质粒同时用限制性内切酶NcoI和XhoI进行双酶切,产物回收后进行酶连,转化后得到质粒pHY-mtnNasRNA-2(pHYmtnNasRNA-3),所用的引物如表3所示。
电转化:按1.3.2节方法制备B. amyloliquefaciensHZ-12的感受态细胞,将抽提得到的表达质粒pHYmtnNasRNA-1、pHY-mtnNasRNA-2和pHY-mtnNasRNA-3以及空载体pHY300PLK分别电转化感受态细胞,涂布Tet抗性平板,37 ℃培养12~16 h得到转化子,取一定数量的转化子分别进行菌落PCR验证,引物用pHY300-F和pHY300-R。
1.3.4 发酵培养条件
从超低温冰箱中取出保藏的菌种划线LB平板培养基中,37 ℃培养基12 h。挑取适量菌体于装有50 mL LB液体培养基的250 mL三角瓶,37 ℃、180 r/min培养9 h(反义RNA抑制mtnN基因表达工程菌种子培养基中加 8 μg/mL Tet抗生素)。吸取1.5%菌液接种于装有25 mL发酵培养基的250 mL三角瓶,37 ℃、180 r/min发酵培养60 h,每个菌设置3 个摇瓶重复。
1.3.5 SAM的高效液相色谱检测
取发酵液0.5 mL,加1.5 mL 0.4 mol/L高氯酸提取1 h(每15 min振荡一次),10 000 r/min离心5 min。取800 μL上清液,加100 μL 2 mol/L NaOH溶液调pH值。利用高效液相色谱检测,色谱柱为XDB-C18(4.6 mm×150 mm,5 μm),检测器为紫外检测器,流动相为甲醇与40 mmol/L NH4H2PO2-2 mmol/L庚烷磺酸钠(18∶82,V/V),流速0.8 mL/min,进样量20 μL,柱温30 ℃,检测波长254 nm。
1.4 数据处理
每组实验设计3 个重复,采用SPSS 20.0进行数据统计分析,评估数据差异的显著性,计算±s,采用 Origin 8.5进行图表绘制。
2 结果与分析
2.1 mtnN基因缺失对解淀粉芽孢杆菌SAM的影响
2.1.1 mtnN基因缺失菌株的构建
利用1.3.2节方法构建重组载体T2(2)-oriΔmtnN,利用表3中相应的引物扩增构建T2(2)-oriΔmtnN的上下游同源臂,上下游同源臂大小分别为776 bp和784 bp,二者经SOE-PCR融合后的产物大小为1 560 bp,上下游同源臂SOE-PCR融合片段与T2(2)-ori质粒经酶切和酶连后转化至E. coli DH5α,选取验证正确的阳性转化子,抽提质粒并用BamHI和XbaI酶切质粒,电泳检测结果如图2所示,重组质粒酶切片段大小与预期相符合,构建好的敲除质粒测序结果显示正确,表明敲除质粒T2(2)-oriΔmtnN构建成功。
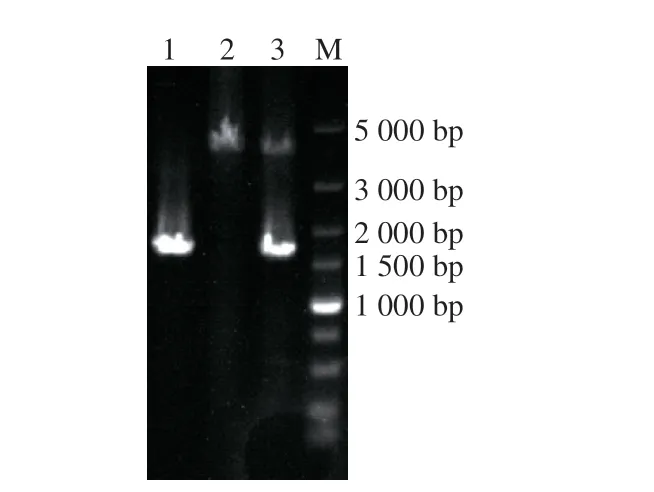
图 2 重组质粒T2(2)-oriΔmtnN双酶切验证Fig. 2 Identification of recombinant plasmid T2(2)-oriΔmtnN by double enzymatic digestion
将已构建完成的重组质粒T2(2)-oriΔmtnN电转化至B. amyloliquefaciensHZ-12中,筛选出阳性转化子,并按照1.3.2节的方法进行单交换和双交换筛选突变株。经菌落PCR验证正确的双交换菌株,抽提其基因组DNA,并用ΔmtnN-YF和ΔmtnN-YR分别验证阳性交换突变株和原始菌株B. amyloliquefaciensHZ-12,片段大小分别为1 965、2 681 bp,PCR验证电泳图如图3所示,回收阳性交换突变株PCR产物经测序确认碱基正确无误。将构建成功的缺失mtnN基因的菌株命名为HZ-12ΔmtnN。
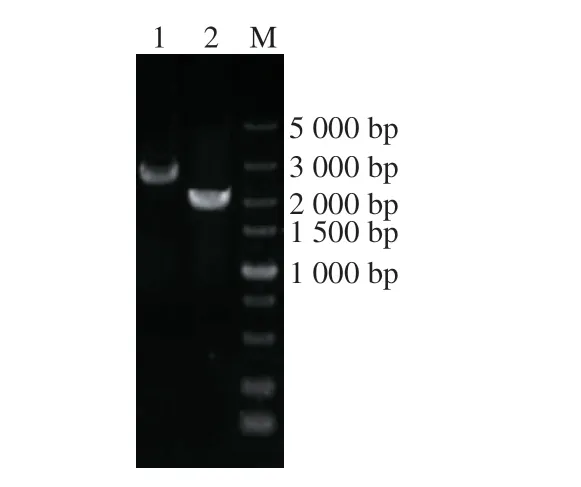
图 3 HZ-12ΔmtnN菌株的PCR电泳图Fig. 3 Electrophoretogram of PCR-amplified products from strain HZ-12ΔmtnN
2.1.2 mtnN基因缺失对SAM含量的影响
为考察mtnN基因的缺失对B. amyloliquefaciens HZ-12合成SAM的影响,按照1.1.3节和1.3.4节发酵培养基和培养条件对工程菌株HZ-12ΔmtnN进行发酵验证,发酵周期60 h。发酵结束后取样检测SAM产量和菌体生物量(OD600nm),结果如图4所示。
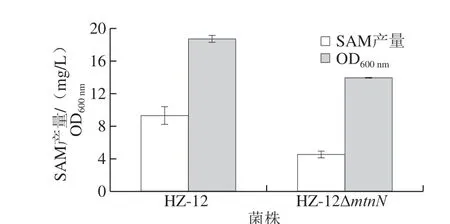
图 4 缺失mtnN基因对SAM产量及菌体生物量的影响Fig. 4 Effect of mtnN knockout on SAM yield and biomass
由图4可以看出,工程菌HZ-12ΔmtnN的生物量相比于原始菌HZ12降低了26%,说明SAHN对菌体的生长有一定的作用,缺失后菌体的生长变缓。此外,工程菌 HZ-12ΔmtnN的SAM产量仅有4.54 mg/L,与原始菌HZ12相比降低了51%,这可能与菌体生长受阻有关。
2.2 反义RNA抑制mtnN基因表达对解淀粉芽孢杆菌SAM的影响
2.2.1 反义RNA抑制mtnN基因表达工程菌的构建
为在不影响菌体生长繁殖的条件下,探索SAHN对B.amyloliquefaciensHZ-12合成SAM的影响。利用反义RNA技术抑制mtnN基因的表达,考察抑制SAHN对B. amyloliquefaciensHZ-12 SAM合成的影响。由于反义RNA的抑制效率与反义RNA在目标基因5’端不翻译区域结合位置有关,因此在mtnN基因5’端不翻译区域选择了不同的结合区域,结合长度分别为100、77 bp和144 bp,而基因编码区域选择的结合长度为100 bp。利用1.3.3节方法构建重组载体pHY-mtnNasRNA-1、pHYmtnNasRNA-2和pHY-mtnNasRNA-3。以B. subtilis 168的基因组为模板,根据表3引物通过PCR扩增启动子P43,大小为305 bp,通过引物mtnNasRNA-F和rrnB-R,以质粒pUC19-mtnNasRNA为模板扩增目的片段和rrnB终止子,大小分别为200、250 bp,经SOE-PCR融合后的产物大小为755 bp,SOE-PCR融合片段与质粒pHY300经酶切和酶连后转化至E. coli DH5α,转化后得到质粒pHYmtnNasRNA-1。通过引物mtnNasRNA-F和mtnNasRNA-2-R (mtnNasRNA-3-R),以质粒pHY-mtnNasRNA-1为模板扩增,大小分别为177、244 bp,产物回收纯化后与pHY-mtnNasRNA-1质粒经酶切和酶连后转化至 E. coli DH5α。选取验证正确的阳性转化子,抽提质粒并用BamHI和XbaI酶切质粒,电泳检测结果如图5所示,重组质粒酶切片段大小与预期相符合,构建好的质粒测序结果显示正确,表明pHY-mtnNasRNA-1、 pHY-mtnNasRNA-2和pHY-mtnNasRNA-3质粒构建成功。
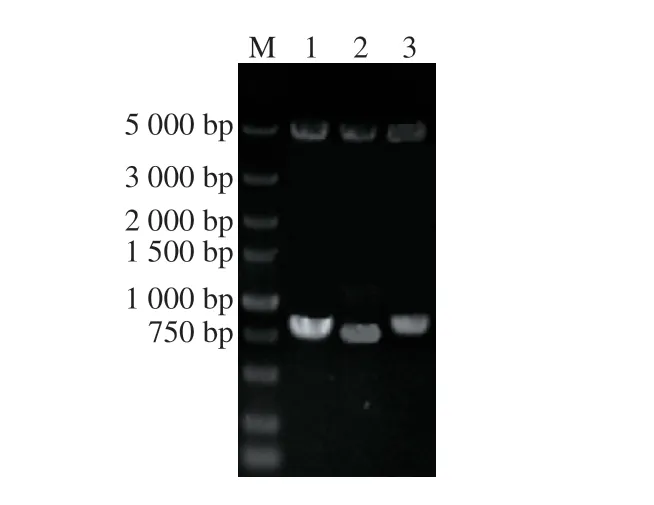
图 5 重组质粒双酶切验证Fig. 5 Identification of recombinant plasmids by double enzymatic digestion
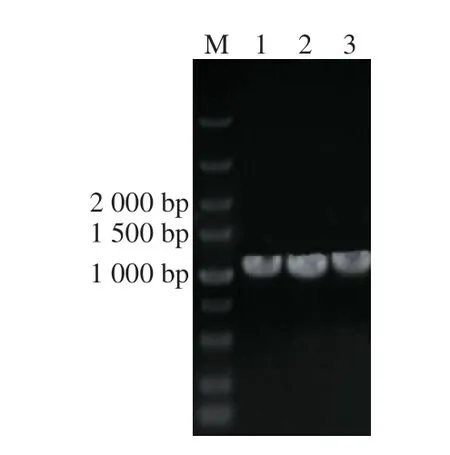
图 6 重组菌株质粒PCR验证Fig. 6 PCR validation of plasmids from recombinant strains
将已构建完成的重组质粒pHY-mtnNasRNA-1、pHY-mtnNasRNA-2和pHY-mtnNasRNA-3电转化至B. amyloliquefaciensHZ-12中,用引物pHY-F/pHY-R进行菌落PCR验证,挑选正确的阳性克隆子进行质粒DNA提取,通过PCR验证(图6),并测序进一步验证,确认序列正确无误,说明反义RNA抑制mtnN基因表达工程菌HZ-12/pHY-mtnNasRNA-1、HZ-12/pHY-mtnNasRNA-2和HZ-12/pHY-mtnNasRNA-3构建成功。
2.2.2 反义RNA抑制mtnN基因表达对SAM含量的影响
研究表明,反义RNA能一定程度抑制某特定基因的表达,而菌体的生长繁殖不受影响。为验证反义RNA抑制mtnN基因对B. amyloliquefaciensHZ-12合成SAM的影响,按照1.1.3节和1.3.4节发酵培养基和培养条件对工程菌HZ-12/pHY-mtnNasRNA-1、HZ-12/pHY-mtnNasRNA-2和HZ-12/pHY-mtnNasRNA-3以及对照菌株HZ-12/pHY300和原始菌HZ-12进行发酵验证,发酵周期60 h。并测定发酵终点的SAM产量和菌体生物量(OD600nm)。
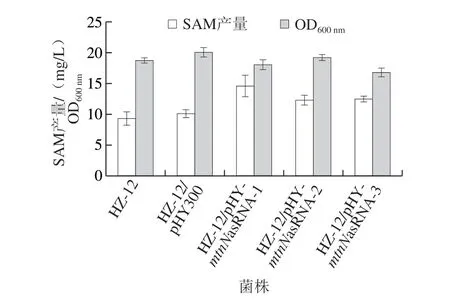
图 7 反义RNA抑制mtnN基因表达对SAM含量的影响Fig. 7 Effect of mtnN expression inhibition by asRNA on SAM production
由图7可以看出,工程菌HZ-12/pHY-mtnNasRNA-1、HZ-12/pHY-mtnNasRNA-2和HZ-12/pHY-mtnNasRNA-3发酵后SAM产量分别为14.58、12.27 mg/L和12.49 mg/L, 相比于对照菌HZ-12/pHY300分别提高了44%、22%和24%,其中工程菌HZ-12/pHY-mtnNasRNA-1效果最明显。由于反义RNA的抑制效率与反义RNA在目标基因5’端不翻译区域结合位置有关,所以在mtnN基因5’端不翻译区域选择不同的结合区域,对mtnN基因的抑制程度不同,从而影响SAM循环途径,导致不同的SAM产量。而工程菌的生物量与对照菌相比没有明显差别,说明反义RNA抑制mtnN基因的表达既可促进SAM的积累,又不影响菌体的生长。
3 讨 论
SAM是人体必需的生物活性辅因子,广泛参与生物体内各种代谢反应,尤其是作为甲基供体参与生物体内一百多种甲基转移酶催化反应[25-26]。mtnN基因同时参与SAM的两个循环利用途径,敲除或抑制该基因有望降低SAM的循环利用,促进SAM的积累。本研究基于基因敲除技术和反义RNA特异性抑制技术,成功构建了mtnN基因缺失菌株HZ-12ΔmtnN以及抑制mtnN的反义RNA工程菌HZ-12/pHY-mtnNasRNA-1、HZ-12/pHY-mtnNasRNA-2和 HZ-12/pHY-mtnNasRNA-3。通过发酵验证,发现缺失mtnN基因的菌株生长受到严重抑制,这与其他研究结果一致,SAHN在细胞内具有重要作用,其活性的下降会造成胞内SAH的累积,SAH是SAM依赖甲基转移酶的反馈抑制剂,胞内低浓度的SAH的积累就会使甲基转移反应得到全面抑制,从而严重影响细菌的生长和分裂[27]。同时,缺失菌株HZ-12ΔmtnN SAM产量仅有4.54 mg/L,与原始菌HZ12相比降低了51%,这可能是其菌体生长受阻造成的直接结果。
传统的基因敲除方法可能会导致菌体生长受缓甚至死亡,而反义RNA介导的调控机制可以被用于代谢途径的微调,常用于平衡细胞生长和产物的合成[28-30]。同样,本研究所构建的反义RNA抑制mtnN基因工程菌株,对菌体的生长没有显著的抑制作用。同时,工程菌的SAM产量相比于对照菌均有显著的提高,特别是工程菌 HZ-12/pHY-mtnNasRNA-1,SAM的产量提高了44%。本研究通过反义RNA抑制技术精细调控mtnN基因,在不影响菌株生长的条件下,显著增强了SAM的合成,为SAM生产菌的代谢工程育种提供了一种新型的策略。总体而言,目前从头合成SAM的产率普遍较低。究其原因可能是目前微生物自身合成甲硫氨酸的能力普遍较弱,未来的研究需通过系统代谢工程改造强化菌株自身合成甲硫氨酸的能力,进一步提高SAM的产量。
猜你喜欢
中国生物化学与分子生物学报(2022年7期)2022-09-07
当代水产(2022年1期)2022-04-26
岭南现代临床外科(2022年1期)2022-03-16
中国药科大学学报(2021年6期)2021-12-31
小学生学习指导·低年级(2021年6期)2021-09-10
考试与评价·七年级版(2021年4期)2021-08-14
食品与发酵工业(2021年7期)2021-04-27
小学阅读指南·低年级版(2018年5期)2018-11-02
中国调味品(2017年2期)2017-03-20
创新科技(2014年12期)2014-07-27
