
肿瘤中甲硫氨酸代谢及其相关基因的表达调控
2022-09-07 05:28赵祎,王萌,杨洋
中国生物化学与分子生物学报 2022年7期
赵 祎, 王 萌, 杨 洋
(北京大学基础医学院生物化学与生物物理系, 北京 100191)
在肿瘤发生过程中,肿瘤细胞常发生一系列细胞代谢变化,称为代谢重编程。甲硫氨酸(methionine)是人体必需氨基酸之一,其功能多样,除参与蛋白质合成外,还可参与一碳单位代谢、叶酸循环,以及多胺、谷胱甘肽、半胱氨酸和核苷酸等多种物质合成。在肿瘤细胞的生长代谢中,甲硫氨酸有着重要作用。1959年,Sugimura等[1]对携带恶性肿瘤的大鼠分别喂食缺乏某种必需氨基酸的饮食,发现甲硫氨酸、异亮氨酸和缬氨酸缺乏可明显抑制大鼠体内肿瘤生长。1976年,Hoffman等[2]发现,大鼠恶性肿瘤细胞及2种转化的人类细胞系均在细胞培养过程中表现出对外源性甲硫氨酸的依赖性,这种特性被称为Hoffman效应。自此之后,肿瘤细胞中的甲硫氨酸代谢重编程被大量研究,在肝癌、乳腺癌、结直肠癌等多种肿瘤细胞中,发现甲硫氨酸代谢的变化以及甲硫氨酸代谢相关酶基因表达的改变[3-6]。本文将从甲硫氨酸代谢出发,总结肿瘤细胞中甲硫氨酸代谢的变化及甲硫氨酸代谢相关酶的基因表达调控,为肿瘤的治疗提供思路。
1 甲硫氨酸代谢
甲硫氨酸是人体必需氨基酸,主要通过食物获得。在细胞中,其主要通过甲硫氨酸循环代谢。甲硫氨酸循环可产生甲基供体S-腺苷甲硫氨酸(S-adenosylmethionine, SAM),参与细胞甲基化反应,并与叶酸代谢、多胺代谢、谷胱甘肽合成、核苷酸合成及转硫途径等多个代谢通路相关,具有广泛的生理功能[7]。在甲硫氨酸循环中,甲硫氨酸在甲硫氨酸腺苷转移酶(methionine adenosyltransferase, MAT)的催化下,与ATP反应生成SAM。SAM是体内最重要的甲基供体,经甲基转移酶(methyltransferase)催化,参与甲基化反应,SAM失甲基生成S-腺苷同型半胱氨酸(S-adenosylhomocysteine, SAH),后者经同型半胱氨酸腺苷转移酶(adenosylhomocysteinase, AHCY)催化失腺苷生成同型半胱氨酸。同型半胱氨酸是甲硫氨酸循环中的重要物质,主要有2个去路,其一可经甲硫氨酸合成酶(methionine synthase, MS)催化,与维生素B12和叶酸循环中的5-甲基-四氢叶酸反应,重新生成甲硫氨酸。同型半胱氨酸可经转硫途径生成半胱氨酸及谷胱甘肽等。此外,甲硫氨酸还可经补救合成途径生成,该途径与多胺代谢密切相关。甲硫氨酸代谢产生SAM参与多胺合成,同时多胺合成副产物甲硫腺苷(methylthioadenosine, MTA)可在甲硫腺苷磷酸化酶(methylthioadenosine phosphorylase,MTAP)催化下重新生成甲硫氨酸,该过程同时有腺嘌呤生成 (见Fig.1)。
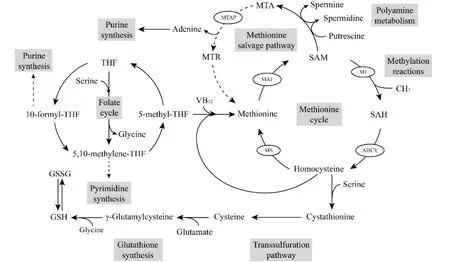
Fig.1 The Methionine cycle and related metabolism pathways In vivo, methionine is metabolized through methionine cycle, which is linked to a variety of metabolic pathways, including the folate cycle, the transsulfuration pathway and the methionine salvage pathway. It affects the nucleotide synthesis, glutathione synthesis, methylation reactions and polyamine metabolism. MAT, methionine adenosyltransferase; SAM, S-adenosylmethionine; MT, methyltransferase; SAH, S-adenosylhomocysteine; AHCY, adenosylhomocysteinase; MS, methionine synthase; VB12, vitamin B12; THF, tetrahydrofolate; GSH, reduced glutathione; GSSG, oxidized glutathione; AMD1, adenosylmethionine decarboxylase 1; MTA, methylthioadenosine; MTAP, methylthioadenosine phosphorylase; MTR, methylthioribose
2 肿瘤的甲硫氨酸依赖性
肿瘤生长对甲硫氨酸依赖性的相关研究,最早见于1959年Sugimura等[1]报道,其发现了饮食中的甲硫氨酸限制能够抑制大鼠体内肿瘤生长。肿瘤细胞增殖对外源甲硫氨酸的依赖性由Hoffman等[2]在1976年发现,其在研究中发现细胞培养过程中的甲硫氨酸由同型半胱氨酸替代后,肿瘤细胞出现增殖受限。自此之后,出现了大量关于肿瘤细胞对于甲硫氨酸依赖性的研究。目前,常将肿瘤细胞对外源性甲硫氨酸的依赖性称为Hoffman效应。
Hoffman效应的相关机制,一直存在诸多假设。早期发现正常细胞在甲硫氨酸缺陷时可出现甲硫氨酸合成酶活性升高,而肿瘤细胞中无此现象[8]。因而认为,Hoffman效应源于肿瘤细胞中同型半胱氨酸转化为甲硫氨酸的障碍。但Hoffman等研究发现,在类似条件下,肿瘤细胞和正常细胞具有相似的内源性甲硫氨酸生成速率,提示肿瘤细胞中的外源性甲硫氨酸和内源性甲硫氨酸存在本质区别,但相关机制尚不明确[9]。此外,也有观点认为,相关依赖性是由于同型半胱氨酸转化途径的改变。在乳腺癌MDA-MB468细胞系的研究中发现,在甲硫氨酸被同型半胱氨酸替代的条件下,同型半胱氨酸在肿瘤细胞中主要经转硫途径代谢,较少参与SAM生成,细胞生长受限依然存在[10]。此时,给予外源性SAM能够解除细胞的生长受限。该效应与甲基化反应、细胞周期阻滞等机制相关[11, 12]。此外,PIK3CA(phosphatidylinositol-4,5-bisphosphate 3-kinase catalytic subunit alpha)基因的突变也可参与肿瘤甲硫氨酸依赖性的形成,其作用机制为该基因突变抑制胱氨酸-谷氨酰胺逆向转运体,减少细胞胱氨酸摄取,从而代偿性加强半胱氨酸经转硫途径代谢[4]。近期又有研究发现,甲硫氨酸能够抑制肿瘤干细胞的细胞自噬,其可能成为Hoffman效应的又一机制[13]。
肿瘤对外源性甲硫氨酸的依赖性还可发生逆转。在甲硫氨酸缺乏的条件下,培养具有外源性甲硫氨酸依赖性的肿瘤细胞,经一段时间可筛选出甲硫氨酸非依赖细胞,这些细胞常丢失肿瘤相关特征,例如非锚定依赖性生长等[14],但其机制尚不明确,可能与染色体异常或表观遗传学改变相关。
3 肿瘤中甲硫氨酸代谢相关基因的表达调控
肿瘤中的甲硫氨酸代谢的改变,常伴随甲硫氨酸代谢酶的基因表达异常,其中以MAT相关的基因表达改变及MTAP基因的缺失最为常见。
3.1 甲硫氨酸腺苷转移酶
3.1.1 MAT概述 MAT催化甲硫氨酸与ATP反应,是SAM生成的关键酶。在人体细胞中,MAT由MAT1A、MAT2A及MAT2B三个基因编码。前两者编码催化活性亚单位MATα1和MATα2,常形成同源二聚体或同源四聚体;后者编码调节亚单位MATβ,MATα与MATβ结合可形成MATαβ复合体[15]。近期研究发现,MATβ主要有2种剪切方式,形成MATβV1和MATβV2,二者均可与MATα2结合,形成复合体[16, 17]。此外,也有MATβV2a和MATβV2b剪切形成复合体[17](见Fig.2)。

Fig.2 MAT enzymes consisted of different oligomers of MAT gene expression products MAT is encoded by MAT1A, MAT2A and MAT2B. MAT1A and MAT2A encode catalytic subunit MATα1 and MATα2, which can form homodimers and homotetramers. MAT2B encodes regulatory subunit MATβ with four splice variants, MATβV1, MATβV2, MATβV2a, MATβV2b. MATβV1 and MATβV2 can combine with MAT (α2)4, forming two kinds of MATα2β, MAT (α2)4(βV1)2 and MAT (α2)4(βV2)2
在人体内,MAT1A主要在肝细胞中表达,在胆管上皮细胞及胰腺腺泡细胞中也有表达[18, 19]。MAT2A在肝内非实质细胞内及所有肝外组织中表达。MATβV1可见于肺、脑、甲状腺、肾上腺、前列腺及婴儿肝组织,而MATβV2在骨骼肌和心血管中表达[17]。在细胞中,MAT最初定位于细胞质,参与SAM生成,随后发现其可在细胞核中分布,参与组蛋白H3K27甲基化[20]。此外,在线粒体基质中,MATα1可抑制细胞色素P4502E1(CYP2E1)的表达,调控线粒体功能[21]。
MAT不同亚型之间的催化活性存在差异。总体而言,MATα1比MATα2活性更高,二者活性差异与其对底物调控作用的反应性相关。对于甲硫氨酸和ATP的米氏常数Km,均有MATα1大于MATα2[22]。此外,SAM作为MAT反应的产物,对MAT活性存在抑制作用,且对于不同种类的MAT,其50%抑制浓度(IC50)存在差异,MATα2的IC50最小,为60 μmol/L,与生理情况下肝内SAM浓度接近;而MATα1的IC50为400~500 μmol/L[23]。综上所述,MATα1相较于MATα2,催化能力更强,且更不易受产物的抑制,因而具有更高的活性。MATα1较高的催化活性差异对于细胞内SAM稳态的维持和肝内SAM的生成至关重要。
3.1.2 肿瘤中的MAT1A、MAT2A基因表达紊乱 在肿瘤中,MAT1A和MAT2A的表达常发生紊乱,表现为MAT1A表达的下调及MAT2A表达上升,被称为MAT1A/MAT2A转换。MAT1A/MAT2A转换可引起细胞内MAT酶总体活性降低,甲硫氨酸代谢能力下降,细胞内SAM生成减少。在肝硬化进展及肝癌发生过程中,这种基因表达紊乱尤为常见。在肝硬化病人中,这种效应可能是引起病人高甲硫氨酸血症的原因之一[24];在肝细胞癌中,MAT1A/MAT2A比例改变可影响DNA甲基化水平,与细胞生长、基因组不稳定性负相关,其降低提示肿瘤恶性程度高[25]。
近年来,MAT1A和MAT2A基因表达改变在肝外组织肿瘤中的作用也逐渐得到重视。在结直肠癌中,有研究发现,MAT1AmRNA表达的升高,提示预后不良[26];在乳腺癌中,MAT1A和MAT2A表达上调,提示肿瘤侵袭性较强,预后不佳[27];MAT1A还被认为与乳腺癌放疗的皮肤反应严重程度相关[28];在膀胱癌中,有研究发现,MAT1A上调参与膀胱癌对化疗药物的抗性形成[29]。在一项细胞研究中,MAT1α与蛋白14-3-3ζ的相互作用与鼠胆管癌及肝癌发生相关,并可促进肝癌细胞的转移和侵袭[30]。
MAT1A和MAT2A基因表达改变的原因复杂,涉及基因表达的各个环节,SAM也被认为作为甲基供体参与MAT1A/MAT2A比例的转换与维持[31]。本文将集中于基因转录和转录后调控,介绍MAT1A和MAT2A基因表达在肿瘤中的改变。
3.1.3MAT1A的基因表达调控MAT1A基因的启动子区域含有多个转录因子的结合区域,可与肝细胞核因子(hepatocyte nuclear factor, HNF)、CCAAT增强子结合蛋白(CCAAT enhancer binding protein, C/EBP)、c-Myc以及糖皮质激素等结合[32]。除作为转录因子参与MAT1A基因表达调控外,C/EBP-β表达还可被短发卡RNA抑制,降低MAT1A启动子活性,下调MAT1A基因表达[33]。c-Myc参与转录调节的机制在近年被阐明。研究者发现,在胆管癌细胞中,c-Myc,转录因子MafG(muscular aponeurotic fibrosarcoma transcription factor G)及c-Maf(cellular muscular aponeurotic fibrosarcoma)过表达,结合于MAT1A启动子抑制性E-box元件,下调MAT1A表达[18]。此外,Prohibitin1(PHB1)可在肝细胞中和MAX形成异二聚体,抑制c-Myc、MafG及c-Maf表达,从而抑制E-box,上调MAT1A。在肝细胞癌和胆管细胞癌中,PHB1表达常下调[34]。
在mRNA水平,MAT1A基因表达主要受非编码RNA及RNA结合因子的调控。富含AU的RNA结合因子(AU-rich RNA binding factor, AUF1)可与MAT1AmRNA的3′非翻译区(3′-untranslated region,3′-UTR)结合,降低mRNA稳定性,下调MAT1A基因表达。在体外诱导大鼠肝细胞去分化,可见AUF1表达上调。在人肝癌细胞中,有AUF1高表达,其敲低可使MAT1AmRNA水平升高。这些研究结果显示,AUF1与MAT1A基因表达存在密切关联[35]。在人肝细胞癌中,miRNA-485-4p、miRNA-495及miRNA-664可诱导癌蛋白LIN28B高表达,抑制抑癌基因Let-7的功能,并下调MAT1A基因表达;MAT1A表达下调可引起SAM合成减少。LIN28B启动子区低甲基化,引起LIN28B表达升高,形成正反馈[36]。一项近期研究发现,lncRNA LINC00662可引起MAT1AmRNA水平下调,及AHCY蛋白水平下调,参与细胞基因组低甲基化形成及肝癌进展[37]。
MAT1A表达下调的影响是可恢复的。在Huh7细胞系中,MAT1A基因的过表达能够稳定地促进SAM水平升高,诱导抑癌基因表达,下调血管生成相关基因的表达,抑制细胞生长并促进其凋亡[38]。这为MAT1A作为靶向治疗的靶点提供了支持。
3.1.4MAT2A的基因表达调控 与MAT1A类似,MAT2A表达也受到多种转录因子调控。肿瘤坏死因子-α(tumor necrosis factor-α, TNF-α)可通过NF-κB和启动子区AP-1元件,诱导MAT2A启动子,促进基因转录[39]。与此类似,转化生长因子β1(transforming growth factor β1, TGF-β1)也可经NF-κB诱导启动子活性,促进肝星状细胞MAT2A基因表达[40]。近期有研究发现,异染色质蛋白1(heterochromatin protein, HP1)参与MAT2A基因表达调控。在HepG2细胞中灭活HP1α,可促进细胞增殖,该效应与MAT1A/MAT2A比例改变有关。在HP1α缺失的细胞中重新诱导HP1α表达,可恢复细胞MAT2A水平,该效应与HP1α参与MAT2A启动子调控有关[41]。
过氧化物酶体增殖物激活受体(peroxisome-proliferator activated receptors, PPAR)参与肝星状细胞MAT2A基因表达调控。PPAR的2个亚型PPARβ和PPARγ在MAT2A启动子中占据相同位点,但其效应相反,PPARγ下调MAT2A转录,而PPARβ诱导其转录;在肝纤维化进展过程中,可有PPARβ表达上调,引起肝星状细胞MAT2A表达相应升高[42]。
人RNA结合蛋白(human RNA-binding protein, HuR)可调节MAT2AmRNA的稳定性。HuR存在甲基化形式methyl-HuR,二者共同调节mRNA稳定性,HuR可提高mRNA稳定性,methyl-HuR可降低mRNA稳定性;在肝细胞去分化及肝细胞癌中,HuR被诱导表达增加,而methyl-HuR减少,这种变化可引起HuR与MAT2AmRNA结合增加,mRNA稳定性增强[35]。
miRNA可抑制MAT2A表达。在HepG2细胞中,miRNA-21-3p可下调MAT2A和MAT2BmRNA的稳定性,抑制细胞生长,引起细胞凋亡,miRNA-21-3p可在药物诱导下产生。此外,具有肿瘤抑制效应的miRNA-34a及miRNA-34b可直接作用于MAT2A,降低其表达,但在包括肝细胞癌在内的多种肿瘤中表达下调[43]。
3.2 甲硫腺苷磷酸化酶
MTAP是甲硫氨酸补救合成途径中甲硫腺苷转化为甲硫核糖(methylthioribose, MTR)的关键酶,其参与嘌呤合成,其基因与表达肿瘤抑制因子p16的CDKN2A基因在位置上非常接近,二者常同时发生缺失。然而,有研究发现,除与CDKN2A基因共同缺失外,肿瘤中也存在MTAP启动子高甲基化及自身的纯合缺失,引起MTAP基因表达的改变[44],提示MTAP的表达与肿瘤发生存在相关性。
在肿瘤细胞中,MTAP的缺失影响广泛。作为MTA代谢酶,其缺失可引起MTA在细胞中的堆积[45]。此外,MTAP可作为肿瘤代谢缺陷的调节因子发挥作用。部分早期研究发现,MTAP缺失的细胞对嘌呤从头合成通路抑制剂的敏感性升高[46]。近期有研究发现,MTA对蛋白精氨酸N-甲基转移酶5(protein arginine N-methyltransferase 5, PRMT5)的SAM结合区域有较高亲和力,故MTA可抑制PRMT5的活性,而其活性对于某些MTAP缺陷细胞系细胞的生长是必需的[45, 47];在MTAP正常表达的细胞中,给予MTA补充,也可介导细胞对PRMT5受抑制的敏感性升高,这提示MTAP缺失对细胞代谢缺陷敏感性的影响与MTA堆积相关。在MTAP缺失肿瘤细胞中,有研究发现,MAT2A缺失可导致PRMT5活性降低,引起肿瘤细胞生长受限。该效应与SAM生成减少、PRMT5甲基化水平降低相关[48];另一研究发现,应用MAT2A抑制剂可干扰PRMT5依赖的mRNA剪接,并介导肿瘤DNA损伤,揭示了PRMT5活性下调抑制肿瘤生长的机制[49]。尽管MTAP缺失在肿瘤细胞中较为多见,但其是否应当作为靶向治疗靶点尚存争议。有研究发现,MTAP缺失无法预测细胞对甲硫氨酸、丝氨酸、胱氨酸限制的反应性,且仅通过甲硫氨酸限制即可清除细胞中MTA堆积,使其与表达MTAP的细胞水平一致[50];在人体胶质母细胞瘤中,MTAP的纯合缺失并不引起体内MTA堆积,这与基质细胞表达的MTAP发挥代谢功能相关,提示了体内外细胞代谢的差异[51]。这些研究使得靶向治疗的必要性存疑。也有研究报道,PRMT5抑制剂能够抑制CD8+T细胞的正常功能,从而抑制机体抗肿瘤的细胞免疫反应[52]。总体而言,MTAP相关的靶向治疗思路是否可行,仍需要更多的研究来论证,以可得到更为准确的结论。
4 甲硫氨酸与肿瘤治疗
根据甲硫氨酸在肿瘤中的代谢特点,目前甲硫氨酸相关的肿瘤治疗方法主要集中于甲硫氨酸限制疗法和靶向治疗两个方向。
4.1 甲硫氨酸限制疗法
从甲硫氨酸的代谢特点来看,甲硫氨酸限制疗法的理论依据主要有两点。首先,甲硫氨酸是人体必需氨基酸,因而甲硫氨酸摄入的限制能够降低体内甲硫氨酸水平。其次,肿瘤细胞对外源性甲硫氨酸存在依赖性,甲硫氨酸的缺乏能够抑制肿瘤生长与增殖。在动物实验中,有研究发现,甲硫氨酸限制疗法对恶性肿瘤的生长和转移具有良好的效果[53]。除单独发挥抗肿瘤作用外,甲硫氨酸还与5-氟尿嘧啶等多种化疗药物存在协同作用[54]。除了限制甲硫氨酸摄入,口服重组甲硫氨酸酶是甲硫氨酸限制疗法的又一方法,并已在晚期前列腺癌、乳腺癌、耐药骨肉瘤、膀胱癌等多种恶性肿瘤治疗中取得进展[55-58]。
尽管甲硫氨酸限制疗法获得一定的研究结果支持,但其距离广泛临床应用还存在诸多问题。一项在小鼠中进行的研究发现,饮食中的甲硫氨酸对于正常肠道菌群的调节是必需的[59]。此外,近期研究发现,T细胞分化及Th细胞正常表观遗传状态的维持需要甲基化反应的参与,因而甲硫氨酸限制可能影响T细胞功能。在一些临床研究中,甲硫氨酸限制引发了较强的不良反应。参与者摄入不含甲硫氨酸的饮食持续24 h,即引起机体的毒性反应[60]。总体而言,尽管甲硫氨酸限制在理论上具有良好的抗肿瘤效应,但在其临床应用过程中,如何避免甲硫氨酸缺乏对机体产生的不可接受的毒性,以及明确甲硫氨酸限制潜在的副作用,都是必须进一步研究解决的问题。此外,也有文献报道,甲硫氨酸增补在细胞研究中亦可降低肝癌细胞侵袭性,这可能为甲硫氨酸的饮食治疗提供新的方向[61]。
4.2 甲硫氨酸靶向治疗
在肿瘤发生过程中,甲硫氨酸代谢酶的基因表达调控常发生各种改变,恢复这些酶的正常功能,有望成为相关肿瘤治疗的又一方案。在此,主要介绍MAT1A、MAT2A和MTAP的相关研究。
由于MAT1A常在肝癌细胞中表达下调,因而目前对于MAT1A靶向治疗的研究,主要集中于解除miRNA对MAT1A表达的抑制。在肝细胞癌模型中发现,作用于miRNA-485-3p、miRNA-495及miRNA-664等3种miRNA的si-RNA可促进MAT1A的基因表达上调,抑制肝细胞癌的生长[36]。
不同于MAT1A,MAT2A的靶向治疗主要集中于抑制蛋白质功能。20世纪70年代,即有学者建议将甲硫氨酸类似物作为一种竞争性抑制剂应用于肿瘤化疗[62]。近年来,相关药物主要有MATα2天然抑制剂及别构调节剂等[63, 64]。近期,MATα2别构抑制剂AG-270在动物体内研究中成功抑制了MTAP缺失的肿瘤的生长,并且耐受性良好,即将开展Ⅰ期临床实验[64]。此外,还有作用于MAT2A基因表达调控的药物报道,IFC-305是一种腺苷衍生物,具有肝的保护作用。研究发现,其可在肝癌细胞中促进MAT1A表达,下调MAT2A表达,改善MAT1A/MAT2A比例。其机制与HuR参与的基因表达调控有关,并最终抑制多胺途径和甲硫氨酸循环,改善肿瘤化疗的效果[65]。
MTAP催化MTA生成甲硫氨酸的过程,伴随着腺嘌呤的合成,其缺失可导致细胞将MTA转化为嘌呤的障碍,因而嘌呤合成是MTAP缺失肿瘤的一个重要治疗靶点,这一治疗思路主要通过应用腺嘌呤类似物抑制核苷酸合成过程的重要中间产物形成[66]。然而,目前该领域尚未在临床研究中取得成功进展,可能还需要更多相关临床前研究,以提供更为精准的药物设计方向。
5 问题与展望
甲硫氨酸作为人体必需氨基酸,具有广泛的生理功能。在肿瘤发生过程中,常伴随甲硫氨酸代谢的紊乱和其代谢通路关键酶基因表达的异常。肿瘤细胞中,甲硫氨酸代谢紊乱常表现为Hoffman效应。Hoffman效应相关的研究为肿瘤的诊断、治疗策略提供了理论基础。甲硫氨酸代谢酶的基因表达异常,主要见于MAT1A和MAT2A,其受到多个方面的影响,包括转录因子、mRNA结合蛋白的调节及非编码RNA的参与等。但各调节通路相互之间的关系尚不够明确,仍需进一步的挖掘。MTAP也常在肿瘤中表达异常,过去认为其与编码肿瘤抑制因子p16的CDKN2A共缺失而发挥作用。而近年来,MTAP独立于CDKN2A的缺失发挥作用也越发得到重视,其效应被认为与MTA的堆积相关,但体内外的研究结果之间仍存在一定的矛盾,体外研究中对其机制的进一步探索和体内研究的验证可能是该领域未来工作的目标。由甲硫氨酸的代谢改变和代谢酶的基因表达异常,分别衍生出两种不同的治疗策略,即甲硫氨酸限制和靶向治疗。然而,这两个治疗思路尚存在各自的问题,诸如甲硫氨酸限制疗法中的短期及长期副作用、靶向治疗中的药物开发和临床试验中遇到的困难,都有待于在未来的研究中进一步解决。总体而言,尽管甲硫氨酸代谢相关的研究距离临床实践仍存在一定的差距,但该领域具有良好研究和应用前景,其在肿瘤的诊断、治疗、预后预测中,均表现出巨大的潜力。
猜你喜欢
中国计划生育和妇产科(2022年5期)2022-11-16
上海师范大学学报·自然科学版(2022年3期)2022-07-11
中国农学通报(2022年13期)2022-05-31
检察风云(2022年5期)2022-04-05
青少年科技博览(中学版)(2022年1期)2022-03-28
文萃报·周五版(2021年38期)2021-09-29
老友(2021年6期)2021-07-01
祝您健康(2020年5期)2020-05-14
健康之友(2020年1期)2020-03-24
健康博览(2020年2期)2020-02-27
