
聚腺苷二磷酸-核糖聚合酶1功能与其抑制剂的作用及耐药机制
2022-09-07 05:28王嘉东
中国生物化学与分子生物学报 2022年7期
刘 雨, 周 筱, 王嘉东
(北京大学基础医学院放射医学系, 北京 100191)
聚腺苷二磷酸-核糖聚合酶1(poly ADP-ribose polymerase-1,PARP1)是PARP(poly ADP-ribose polymerase)超家族中研究最广泛的核酶,其功能众多,且与DNA损伤修复过程密切相关。近年来,以奥拉帕尼(Olaparib)为首的PARP抑制剂被广泛应用于家族性乳腺癌和卵巢癌的维持治疗中。多项研究证实,PARPi能显著提高患者的无进展生存期[1]。然而,有部分乳腺癌基因1/2(breast cancer gene 1/2,BRCA1/2)突变型肿瘤细胞表现出对PARPi的抗性,成为目前PARPi在临床应用上的一大问题。这提示我们对PARP1生理功能及调控因素的认识尚不充足。因此,阐明PARP1参与DNA损伤修复的具体过程,并从中得出肿瘤细胞对PARPi耐药性的合理解释,具有重大医学和社会意义。本文首先对PARP1的功能做一总体回顾,而后围绕PARP1在DNA损伤修复方面的作用,阐述PARP抑制剂的抗肿瘤机制及耐药机制。
1 聚腺苷二磷酸-核糖聚合酶1的结构与功能
PARP1全长1 014个氨基酸,分子量约113 kD,其结构主要可分为N-端的DNA结合结构域(DNA-binding domain)、中部的自修饰结构域(automodification domain)以及C-端的催化结构域(catalytic domain)(Fig.1A)[2]。顾名思义,PARP1具有催化“PAR修饰(PARylation)”反应的酶活性。所谓PAR修饰,是指以NAD+为供体,在底物上连接一串多聚ADP-核糖链(Fig.1B)[3]。近年来,随着人们对PARP1认识的不断深入,发现除经典的PAR修饰外,PARP1也能进行乙酰化、磷酸化、泛素化和类泛素化等多种翻译后修饰,为PARP1功能的多样性奠定了基础[4]。
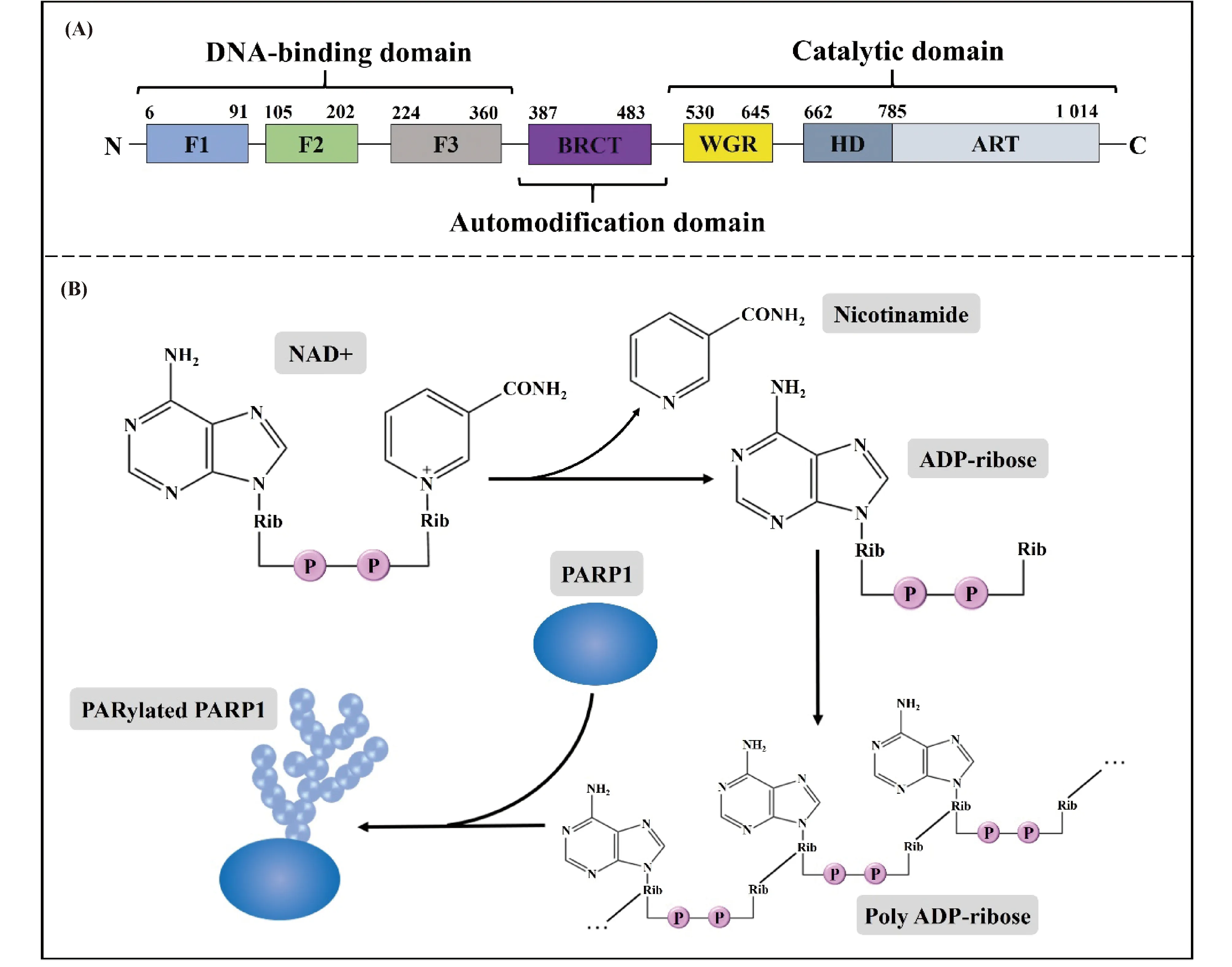
Fig.1 The main structural domain of PARP1 (A) and the catalytic process of PARP1 PARylation (B) PARP1 catalyze the reaction of nicotinamide removing from NAD+ to form ADP-ribose. Multiple ADP-ribose molecules are sequentially connected to PARP1 through glycosidic bonds and finally form PARylated PARP1
1.1 PARP1是DNA损伤修复的感受器
细胞中的DNA断裂损伤主要可分为单链断裂(single strand break,SSB)和双链断裂(double strand break,DSB)两种。在细胞发生SSB或DSB后,PARP1的DNA结合结构域可识别并结合损伤位点,而后催化结构域催化PAR修饰反应。该反应以其自身为底物,将PAR链连接在自身的BRCT结构域上,而后促进DNA损伤修复效应蛋白质募集并结合在PAR链上,启动修复过程[5]。PARP1脱落后,这些修复效应蛋白质在核小体PAR修饰因子1(histone PARylation factor 1,HPF1)帮助下,继续锚定在损伤位点发挥功能[6]。因此,PARP1被形象地称为“DNA损伤修复的感受器”,其中尤以SSB修复效应分子的招募最为重要。
PARP1介导的SSB损伤修复可分为4个步骤(Fig.2A),依次为:识别(lesion detection)、末端加工(end processing)、缝隙填补(gap filling)和连接(ligation)。修复过程中的重要分子已在图中表示,包括提供修复反应平台的X射线交错互补修复蛋白1(X-ray repair cross complementing 1,XRCC1)[7],切除不规则末端的多聚核苷酸激酶/磷酸酶(polynucleotide kinase /phosphatase,PNKP)[8]和脱嘌呤嘧啶核酸内切酶1(apurinic/apyrimidinic endonuclease 1,APE1)[9],切除后端小段完整DNA单链的结构特异性核酸酶1(flap structure-specific endonuclease 1,FEN1)[10],以及相应的DNA聚合酶和连接酶等[11]。
然而,DNA单链损伤并不会直接对细胞表现致死效应,引起细胞凋亡的通常是DNA双链断裂。在细胞有丝分裂的S期,如果一条存在SSB位点的DNA链发生了复制,那么复制叉在进行到损伤位点时会发生停滞,等待PARP1等一系列前文提及的蛋白质对SSB位点进行修复。此时,如果细胞未能完成SSB修复,复制叉将发生崩溃(collapse),导致DSB的形成[12]。细胞修复DSB的途径主要可分为同源重组(homologous recombination,HR)和非同源末端连接(non-homologous end joining,NHEJ)两种。其中,同源重组的保真性强,是细胞修复DSB的最佳途径。但由于HR过程需要与损伤链同源的双链DNA作为模板,因此只出现在S/G2期[13]。HR修复也可分为4个步骤(Fig.2B),分别为末端切除(end resection)、链侵入(strand invasion)、链延伸(DNA synthesis)和霍利迪连接体解离(dissolution of Holliday junction,dHJ)[14-17]。与SSB修复类似,在DSB中,PARP1也参与招募修复效应分子。例如,BRCA1、BARD1和MRN复合体中的MRE11等,但不是这些分子发挥功能的必需条件[18]。
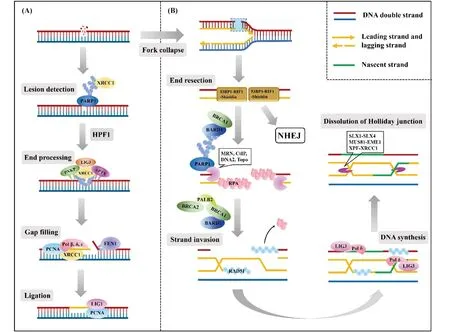
Fig.2 DNA single-strand and double-strand break repair pathways (A) After DNA single strand damage occurs, PARP1 takes the lead in locating and identifying the damage, recruiting following proteins to degrade the damage end and extend nascent DNA. (B) If the function of PARP1 is inhibited, the replication fork will collapse during the S/G2 phase, resulting in double-strand breaks and initiating the HR repair pathway. BRCA1/2 are key molecules in HR pathway. If they are mutant, cells can only choose the NHEJ pathway to repair double-strand damage, which is low fidelity, causing genomic instability and finally cell apoptosis
由此看出,细胞对DNA损伤有完备的应对措施。在损伤初期,PARP1可识别启动SSB修复途径;而即使SSB修复未能完成,也可在后续进行DSB修复,使细胞能够存活。这也是PARPi专一靶向BRCA突变肿瘤细胞,而对正常细胞影响较小的生理基础。
1.2 PARP1与多种蛋白质相互作用维持基因组稳定性
基因组稳定性(genome stability)是指细胞面对复制错误和DNA损伤时,能够修复异常,保证遗传物质在世代传递过程中稳定不变的能力[19]。维持基因组稳定性,一方面需要细胞具有稳定可靠的DNA损伤修复机制;另一方面,也需要细胞中多种机制共同发挥作用,为DNA损伤修复的进行提供有利条件。
复制叉停滞(replication fork stall)为DNA损伤修复过程提供充足的时间。经典理论认为,PARP1在损伤位点的募集在复制叉停滞中发挥主要作用[20],但这无法解释在PARPi导致PARP捕获的情况下,复制叉反而更不稳定。最近的研究认为,PARP1在损伤位点的募集和PAR修饰可抑制DNA修复相关解旋酶RECQ1的活性,而RECQ1激活会导致停滞的复制叉重启[21],明确了PARP1维持复制叉停滞的作用机制。
DNA损伤修复过程需要多种蛋白质的募集,这就需要损伤位点附近具有足够的空间。一般情况下,染色质高度聚集并缠绕在组蛋白上,不利于DNA损伤修复,这就需要损伤位点附近形成一段较为松散的DNA结构[22]。Smeenk等[23]揭示,PARP1可PAR修饰核小体组蛋白并促使其解离。同时,PARP1自身的PAR修饰可以招募多种染色质重塑因子(chromatin remodeling factor,CRF)。例如,ALC1、CHD2、CARM1和SMARCA5等。这些分子可进一步促进核小体解聚,使缠绕的染色质转变为松散的线性结构[24, 25]。
另外,PARP1也能影响DSB修复途径的选择和DNA损伤检查点的激活。此二者机制与PARPi抗肿瘤的作用机制相关,将在后文详细说明。
1.3 PARP1参与神经退行性疾病的发生发展
早年有学者发现,在轻度氧化应激的情况下,PARP1缺陷的组织细胞比正常细胞更易发生凋亡。尽管PARP1在维持基因组稳定性方面的作用可以部分解释这一现象,但仍不够充分[26]。近年来,有越来越多的证据支持PARP1广泛参与细胞自噬、衰老和炎症过程[4]。其在神经退行性疾病中发挥的功能也成为最新研究的热点。
神经退行性疾病的一大病理改变是细胞内的异常蛋白质沉积。其中,许多蛋白质被证实有增强PARP1活性的能力。例如,β-淀粉样蛋白(amyloid β-protein,Aβ)、Tau蛋白和α-突触核蛋白(α-synuclein)等[27-29]。PARP1活性的增强,一方面对这些蛋白质进行PAR修饰,导致其进一步聚集并加重神经元毒性;另一方面,过度的PAR修饰,可使NAD+和ATP耗竭,引起细胞内ROS生成和能量代谢障碍,最终导致细胞死亡[30]。
慢性炎症也是神经退行性疾病的诱因之一。沉默调节蛋白(sirtuin,SIRT)是一类NAD+依赖的组蛋白去乙酰化酶。多项研究证实,SIRT1对神经细胞有营养和保护作用。例如,SIRT1可使Tau蛋白去乙酰化而变得不稳定,以减少Tau蛋白聚集[31]。而PARP1过度激活会抑制SIRT1活性[32],可能的后果包括SIRT1介导的高迁移率族蛋白1(high mobility group box 1,HMGB1)乙酰化水平降低,使HMGB1释放到细胞外,进而发挥与炎症因子类似的作用引起神经炎症[33];以及通过激活NF-κB通路,增强炎症因子,例如肿瘤坏死因子-α(tumor necrosis factor-α,TNF-α)、白介素-1β(interleukin-1β,IL-1β)、环氧合酶2(cyclooxygenase 2,COX2)及CXC亚族趋化因子配体2(CXC subfamily chemokine 2,CXCL2)等的表达[34]。
目前临床上处理剖宫产术后镇痛多采用硬膜外镇痛或静脉镇痛,硬膜外镇痛虽然效果确切但低血压、运动阻滞、尿潴留、抑制胃肠道蠕动发生率高,而静脉镇痛效果差,且容易发生呼吸抑制、过度镇静、恶心呕吐等不良反应[11]。神经阻滞技术是术后多模式镇痛的一种安全有效的补充方法。对阿片类药物反应较差或耐药患者,神经阻滞镇痛效果良好。
总之,PARP1的过度激活在多方面影响细胞正常生理功能,促进神经退行性疾病的发生发展。对此,科研人员发现了一些仅抑制PARP1活性,而不导致PARP捕获的“新型PARP抑制剂”[35],被认为可有望用于神经退行性疾病的治疗。
2 聚腺苷二磷酸-核糖聚合酶抑制剂的抗肿瘤作用机制
PARPi可影响PARP1的功能,使细胞失去修复SSB的能力,导致SSB进展为DSB。对于正常细胞而言,正如上文所述,细胞仍可进行同源重组修复。然而,对于BRCA1或BRCA2基因突变的细胞,其HR功能缺陷,细胞因不能修复DNA损伤,最终走向凋亡。这种由单一因素作用不表现对细胞的杀伤,而二者共同作用下引起细胞死亡的效应,被称为“合成致死(synthetic lethality)”[36]。
2.1 PARPi可抑制PARP1催化活性
前已述及,PARP1进行PAR修饰使用的ADP-核糖供体是NAD+。而奥拉帕尼、卢卡帕尼等PARPi中有与NAD+的烟酰胺类似的环结构(Fig.3)[37],这是PARPi竞争性结合PARP1的结构基础。PARP1结合PARPi后,不再能催化PAR修饰反应,也就不能启动SSB修复通路。
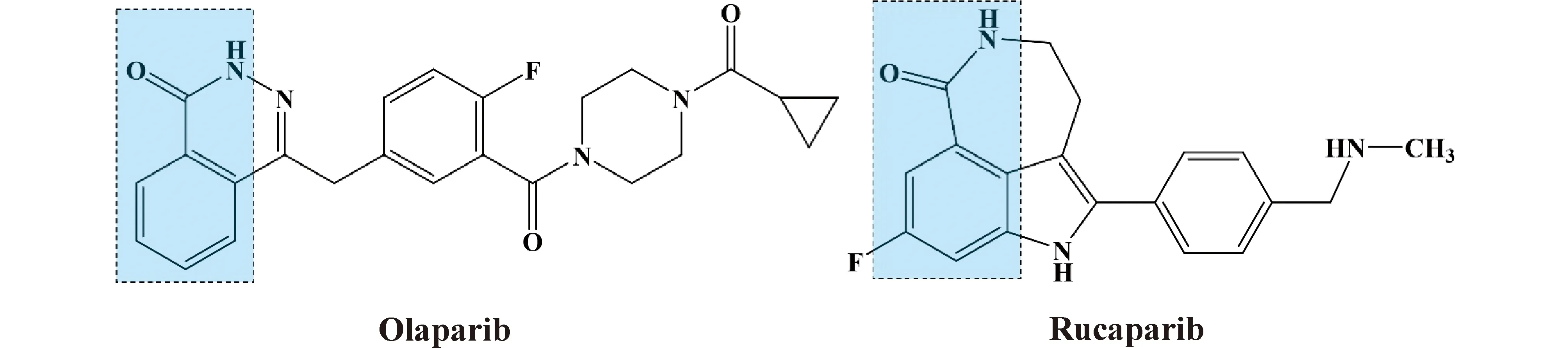
Fig.3 Molecular structure of Olaparib and Rucaparib Nicotinamide-like structures are shown in the dotted box
通过X线晶体衍射技术可以判明,PARPi的类烟酰胺结构可与PARP1形成3个关键的氢键,分别位于Gly863、Ser904和Tyr907[38]。此外,PARPi的特殊结构还会与PARP1形成共价连接,具体结合位点根据PARPi种类而略有不同,常见的位点包括Tyr889、Tyr896、Phe897、Ala898和Lys903等[39-41]。不难看出,这些位点仍属于PARP1的催化结构域。换言之,PARPi与PARP1的结合不会影响其与DNA结合的能力,这对解释PARPi耐药性具有重要意义。
2.2 PARP1捕获是PARPi杀伤肿瘤的主要原因
虽然PARPi抑制PARP1催化活性的能力客观存在,但这种能力尚不能完全解释PARPi的抗肿瘤活性。因为如果仅从抑制PARP1酶活性的角度来看,使用PARPi和敲除PARP1的效果应该是一致的。然而,2012年Murai等[42]发现,PARP1敲除组细胞存活率显著高于使用PARPi组,同时如果敲除PARPi组细胞的PARP1,细胞存活率将提高,表明除抑制PARP1活性,PARPi还有其他抗肿瘤机制。近年的研究表明,相比于PARP1催化活性的抑制,“PARP捕获(PARP trapping)”是PARPi更为重要的作用机制[43]。体外和体内研究均证实,在正常情况下,PARP1发生PAR修饰后,其与DNA结合能力将减弱,导致其自动从损伤位点脱落,以便相关效应酶定位在损伤位点进行修复[44]。然而,在PARPi存在情况下,PARP1不能正确进行PAR修饰,也就不能从损伤位点脱离,即形成“PARP捕获”。导致PARP1不能正常发挥功能的同时,还成为DNA复制和转录过程的障碍,加剧了基因组不稳定。此外,一些肿瘤细胞中存在PARP1基因的突变,而其仍能存活[45],提示PARP1并非SSB修复通路的唯一启动因子。事实上,细胞中还有其他参与修复SSB通路,例如在损伤初期,RPA也可与ssDNA结合,而后募集损伤检查点激酶ATR,ATR发生自身磷酸化,激活后续SSB修复相关酶[46, 47]。PARP1捕获也同时影响了这些分子对SSB的修复作用。
2.3 PARP1其他功能的抑制导致基因组不稳定
前已述及,PARP1参与DSB修复通路的选择。在细胞的S/G2期,理论上HR和NHEJ都有进行的基础,此时细胞选用哪种途径进行修复,实际上是2条通路间的分子竞争的结果。例如,在DSB初期的53BP1-RIF1-Shieldin复合体保护末端,不利于HR途径中MRN复合体对末端的剪切加工,此时如BRCA1活性强,则能去除53BP1,进行HR,而如果53BP1活性强,则进行NHEJ[48]。近年有研究证实,PARP1激活产生的PAR链能直接与NHEJ修复的识别分子Ku70和Ku80结合,抑制二者活性,从而抑制NHEJ途径[49]。因此,在PARPi存在情况下,NHEJ通路的抑制会被解除,与BRCA突变细胞的HR缺陷表型产生协同作用,使细胞选择NHEJ修复途径,而NHEJ的保真性不强,因此常导致基因组不稳定。
PARPi对S期DNA损伤检查点的抑制也是导致基因组不稳定的原因之一。DNAFiber研究发现,在PARPi存在时,复制叉速度提高了约1.4倍[50],复制叉行进速率过快常导致复制错误,无法停滞的复制叉也直接加快了崩溃的速度。一种合理的解释是,在复制叉上存在SSB位点时,PARP1可以PAR修饰p53,从而促进p53活性,进而启动DNA损伤检查点,使复制叉停滞[51, 52],以便损伤得到修复。而PARPi使这些功能丧失,引发复制叉迅速崩溃。
综上所述,PARPi抗肿瘤的作用机制可总结如Fig.4。
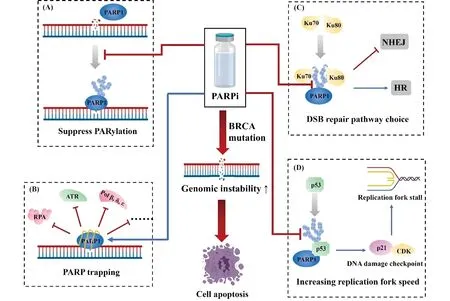
Fig.4 The main anti-tumor mechanism of PARPi PARPi can interrupt cells from repairing DNA single strand damage by (A) competitive binding of PARP1 to suppress PARylation and (B) making PARP1 trapping on DNA break site. In addition, PARPi can also (C) reduce the binding of Ku70 and KU80 to PARP1, thus affecting the choice of DSB repair pathway. PARPi can also (D) interfere with the DNA damage checkpoint by affecting the PAR modification of p53. Together with BRCA deficiency, the above mechanisms lead to cell genomic instability and eventually apoptosis
3 肿瘤细胞对聚腺苷二磷酸-核糖聚合酶抑制剂的耐药机制
耐药肿瘤细胞无一例外地都有一个或多个基因的突变,这些基因从不同层面影响DNA损伤修复过程[53]。从机制上,与耐药相关的突变可分为以下几类。
3.1 HR途径恢复
PARPi抗肿瘤主要依赖合成致死效应,因此细胞自身HR通路的缺陷对PARPi的杀伤作用是至关重要的。BRCA1/2突变是临床最常见的HR缺陷型肿瘤,也是PARPi的用药指征之一。但临床发现,少数患者在接受PARPi治疗后,自身BRCA1/2会发生回归突变。其中,2次移码突变占绝大多数,即原本发生1次移码突变,导致BRCA转录翻译提前终止。而再次突变时,2次相加缺失的碱基数恰为3的倍数,使BRCA基因在一定程度上恢复活性[54]。
在HR恢复表型中占更大比例的是53BP1缺失型[55]。前文提到,细胞面对DSB时存在HR和NHEJ途径的选择机制,53BP1因能保护损伤末端不受HR所必要的剪切加工,可促使细胞倾向选择NHEJ修复途径。而如53BP1缺失,则会导致细胞倾向选择HR,在一定程度上避免了细胞凋亡,致使细胞对PARPi耐药。最新的临床报告证实,与53BP1共同发挥作用的Shieldin基因及其下游的肿瘤蛋白P53基因(tumor protein P53 gene,TP53)突变,也会带来类似的结果[56, 57]。
目前,临床上开发了多种联合用药策略,以应对肿瘤细胞此种类型的耐药,主要思路是通过人为干预HR的其他靶点,减弱细胞HR活性。例如,联用CDK抑制剂、HSP90抑制剂、HDAC抑制剂以及联合电离辐射等[58]。其中,CDK是HR第一步末端剪切的重要活化因子[59],热休克蛋白90(heat shock protein 90,HSP90)是BRCA1发挥活性的辅因子[25]。HDAC是细胞中重要的组蛋白去乙酰化酶,在表观遗传调控中发挥作用。如果去乙酰化作用被抑制,许多与HR修复相关基因的表达将会下调,例如细胞周期检测点激酶1(cell cycle checkpoint kinase 1,CHK1)、RAD51、BRCA1和HSP90等[60, 61]。此外,有实验证实,电离辐射能促进细胞核中BRCA1向细胞质转运,使核内BRCA1耗竭,导致HR缺陷[62]。
3.2 PARP1与SSB位点结合减弱
PARP1捕获是PARPi杀伤肿瘤的主要机制。细胞中如缺失PARP1基因,损伤位点便不会有捕获的PARP1引发复制压力,且细胞也能通过其他途径修复SSB,导致PARPi耐药。但这类突变一般仅在科研工作中人为构建,在临床上尚未发现[63]。更为常见的是PAR糖苷水解酶(poly ADP-ribose glycohydrolase,PARG)基因的缺失[64]。PARG可水解PAR链,与PARP1功能相拮抗。在生理情况下,PARG活性对PARP1功能的维持具有重要意义。PARP1经过自身PAR修饰后,从SSB位点脱落,由PARG去除PAR修饰,使PARP1恢复与DNA的结合能力和PAR修饰催化活性,以便下次发生SSB时行使正常功能[65]。而如果细胞中PARG缺失,一方面PARP1即便被PARPi结合,仍可被其他酶进行PAR修饰,从而正常启动SSB损伤修复通路;另一方面,由于脱离的PARP1不能被及时去除PAR修饰,在之后的SSB过程中不能正常与DNA结合,也就减少了PARP1捕获。对于PARG缺失导致耐药,一些人工合成PAR链降解剂,如iRucaparib-AP6被证实有良好的疗效[66]。
3.3 复制叉稳定性增强
复制叉停滞是细胞在面对复制压力时的保护机制,也是细胞得以修复DNA损伤并继续存活的保证。BRCA1/2除上述HR修复通路的功能外,也参与复制叉停滞的保护,对于BRCA1/2突变细胞,复制叉呈现不稳定性,易被降解[67, 68]。细胞中存在多种降解复制叉的机制,其中具有核酸酶活性的效应分子主要是甲磺酸盐及紫外线敏感基因81(methanesulfonate and ultraviolet sensitive gene clone 81,MUS81)编码蛋白质MUS81和减数分裂重组蛋白11(meiotic recombinant protein11,MRE11)两种。这二者识别和招募的机制略有不同,其中MUS81主要通过甲基化酶Zeste基因增强子(enhancer of zeste homolog 2, EZH2)进行H3K27me3修饰而被招募到复制叉[69],而MRE11主要通过Pax反式激活域相互作用蛋白(Pax transactivation domain-interacting protein,PTIP)-MLL3-MLL4甲基化酶复合体进行H3K4me3修饰而被招募[70]。
因此,细胞如发生EZH2和PTIP基因的缺失,组蛋白相应位点不能被甲基化,因而核酸酶不能被招募到停滞的复制叉附近,抑制了复制叉的降解。复制叉稳定性增强,意味着细胞有更充足的时间修复DNA损伤,使肿瘤细胞在PARPi的攻击下得以存活。针对此类突变的治疗策略主要是通过联用CDK抑制剂、p53抑制剂等[71],促使细胞DNA损伤检查点失效,从而使复制叉加速。
3.4 主动药物外排增加
ABC转运蛋白(ATP-binding cassette transporter),是一种以ATP为驱动能源的跨膜运输蛋白质。ABC转运蛋白可以耗能形式将细胞中代谢产物、毒素和药物转运至膜外。其中,与药物外排相关的蛋白质主要是ABC家族成员中的1a和1b(ABC1a/b)[72]。有研究证实,在PARPi耐药乳腺癌中,ABC1a/b的水平有不同程度的上调(2~85倍)。且在应用2种常见的ABC抑制剂Elacridar和Verapamil后,肿瘤耐药性有所逆转,证实ABC转运蛋白在PARPi耐药中发挥的作用[73]。
事实上,ABC转运蛋白的上调可见于大量肿瘤患者中,且常导致多药耐药性。克服药物外排的方法也并无特异性,主要思路包括联用ABC抑制剂和改进药物结构,使ABC不能外排。ABC抑制剂目前研究比较成熟,已有最新的3代药物,例如Cyclosporine、VX-710、GF120918和XR-9576等[74];而在药物结构的改进方面,发现蒽环结构可极大阻碍ABC转运[75],为新型PARPi研发提供了方向。
PARPi的耐药机制和相关突变基因可总结于Table 1。值得注意的是,除基因突变可导致PARPi耐药,最近的研究发现,PARPi耐药还可能与细胞表观遗传修饰和miRNA的异常表达相关[76, 77]。不难看出,因PARPi耐药的机制各异,在选择对抗PARPi耐药的方案之前,了解患者存在的基因突变,找到具体的耐药机制,对治疗具有较大帮助。
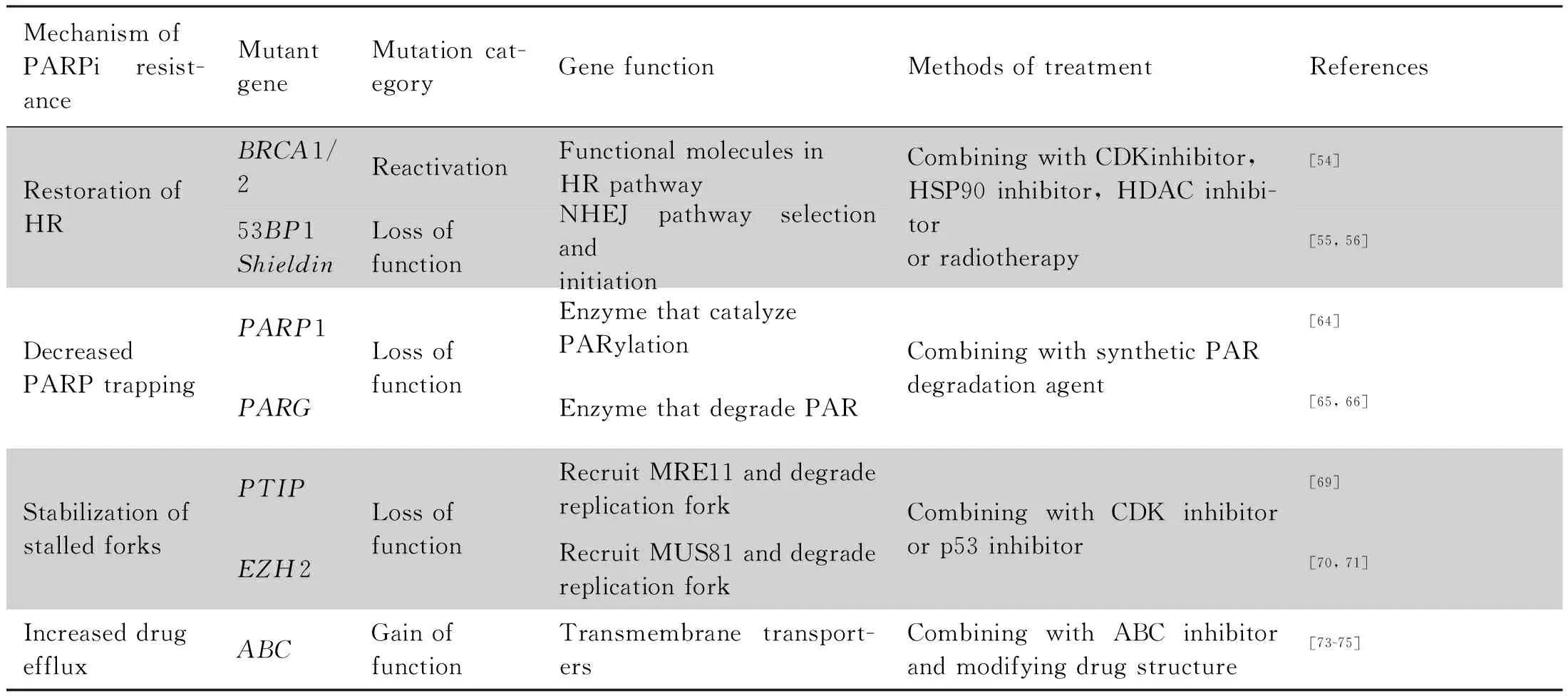
Table 1 Mechanism and related genes of PARPi resistance
4 问题与展望
虽然PARP1参与DNA损伤修复已是较为经典的认识,但该过程仍有诸多细节值得探究。例如,PARP1与DNA的结合-解离过程是否需要其它分子的协助,影响PARP1底物选择和催化活性的分子有哪些,PARPi结构的不同是如何影响PARP1捕获程度的等。解明这些问题,无疑将会为克服PARPi耐药性,研制更为高效的PARPi提供支持。另外,与PARPi相关的基础研究也在不断发展,包括在基因层面寻找与BRCA突变类似的HR缺陷表型肿瘤,从而将PARPi推广到多种类型癌症的治疗中;以及仿照PARPi与HR缺陷的合成致死概念设计药物靶点,开发全新的抗肿瘤用药策略。与此同时,随着研究的进展,人们对PARP1功能的认识已经不再局限在DNA损伤修复上。近年越来越多的研究指出,PARP1在细胞周期和能量代谢调控等方面发挥更为广泛的作用,这让人们认识到PARP1在多种疾病的发生发展中均扮演重要角色。总之,在意识到临床问题,开发新疗法的同时,基础研究也应持续推进,期待有朝一日,我们能对PARP1的功能有全面的认识,PARPi能够得到广泛的应用。
猜你喜欢
中国农业科学(2022年16期)2022-09-19
中国农业科学(2022年15期)2022-08-09
中国典型病例大全(2022年11期)2022-05-13
健康体检与管理(2022年4期)2022-05-13
医学概论(2022年4期)2022-04-24
中国典型病例大全(2022年7期)2022-04-22
中国药房(2022年7期)2022-04-14
电脑报(2020年40期)2020-11-06
保健与生活(2019年2期)2019-08-01
电脑知识与技术(2018年19期)2018-11-01
