
几种氧杂蒽衍生物吸收光谱的理论研究
2020-03-30 08:39赵文伟
山东化工 2020年4期
赵文伟
(临沂大学 化学化工学院,山东 临沂 276005)
氧杂蒽,分子式C13H10O,结构如图1所示,由一个氧及一个碳原子连接着两个苯环的芳环衍生物,碳与氧处于对位,并形成一个中间六元环。氧杂蒽本身并没有特殊的用途,但它是一大类荧光物质和染料(呫吨染料)的基本母体化合物和一些合成药物的基本骨架[1-5]。氧杂蒽环上引入不同的取代基,形成不同的化合物,不仅影响染料的最大吸收波长,也直接影响它们的应用性能。3,6-位氢原子被氨基化合物取代,形成派洛宁化合物,如图1所示。氨基上的氢原子被不同的烷基,对增色或荧光效应都有较大的影响。分子中9-位C原子可以与第三芳环相连,形成罗丹明化合物。本文对派洛宁20、派洛宁B、派洛宁Y和罗丹明123进行理论研究,探讨其结构特点,以及不同氨基和9位C原子上的取代基对吸收光谱波长的影响。

图1 氧杂蒽及其衍生物的结构
1 计算方法
几何优化使用B3LYP方法和6-31G(d,p)基组进行,几何优化均使用Gaussian 09默认的收敛标准。DFT方法对罗丹明6G的优化结构和晶体结构十分接近[7]。极化连续介质模型(polarizable continuum model,PCM)用来模拟溶剂对结构和光谱的影响。在优化的几何结构基础上,使用含时密度泛函理论TD-DFT(BP86泛函)和6-31G(d,p)基组计算吸收波长和振子强度。自然键轨道分析使用NBO3.1程序完成。
2 结果与讨论
2.1 几何结构
无论派洛宁化合物还是罗丹明123,都是左右对称的,对称面穿过C9-O10。派洛宁化合物中两个苯环受到取代基的影响,键长发生变化,但是变化不大,和苯环单体的C-C键键长接近。
派洛宁化合物的一个重要几何特点是C9-C11的键长,在派洛宁20,B,Y中,该键的键长分别是1.3983,1.3977,1.3980埃,几乎和苯环上的C-C共轭键(1.3997埃)相等。说明C9参与了整个氧杂蒽环的大π键共轭。NBO分析证明C9的杂化方式是sp2,与苯环上C原子的杂化方式相同。自然键轨道分析结果列在表1中。自然键轨道分析认为氧杂蒽环上的O原子为sp2杂化,其中两个sp2杂化轨道参与成σ键,一个sp2轨道处在平面上,未参与杂化的p轨道与C原子未参与杂化的p轨道形成大π键。这样氧杂蒽中间的六元环有7个共轭的π电子,O原子贡献两个孤对电子,5个C原子各贡献1个π电子,不符合4n+2的规则,因此没有芳香性。从结构上看,派洛宁的C-N键键长在1.352埃左右,比一般的C-N单键(1.47埃)短,N原子与氧杂蒽环处在同一个平面上。在罗丹明123中,氧杂蒽环的结构与派洛宁的类似,苯酸酐相当于一个吸电子基,苯甲酸酐和氧杂蒽环相连的C-C键键长是1.49696埃,是一个典型的C-C单键。受苯酸酐的影响,氧杂蒽环不再是一个平面结构,两个苯环呈一定的夹角。
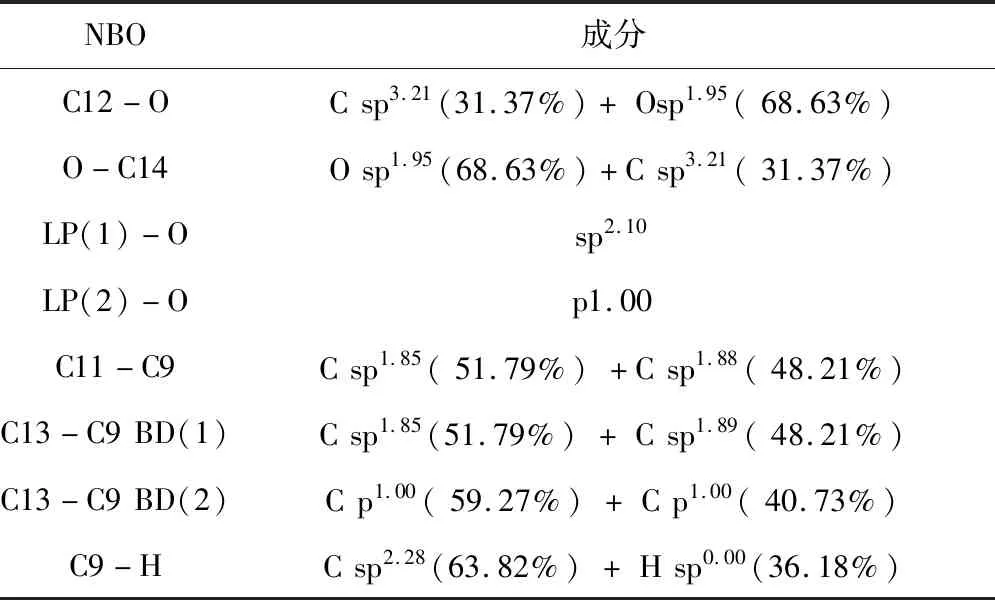
表1 自然键轨道分析结果
2.2 吸收光谱性质
在优化的几何构型的基础上,使用TD-DFT方法分别计算了派洛宁20、B、Y和罗丹明123的从基态到10个激发态的跃迁能量、吸收波长和振子强度(见表2)。其中跃迁强度最强的都是第一激发态,因为其对应的振子强度最大,是实验上观测到的电子跃迁,因此主要讨论电子从基态到第一激发态的电子跃迁。派洛宁20、B和Y从基态到第一激发态的吸收波长计算值分别是487.19nm,514.67nm和511.46nm,与实验值[8-10]十分接近。

表2 在PCM-B3LYP/6-31+G(d,P) 水平上计算的派洛宁20,B和Y的激发能、吸收波长、振子强度和组态
注:aref 8;bref 9;cref 10
罗丹明123分子中,由于受到苯酸酐的静电排斥的影响,氧杂蒽环不再是平面结构的,对称面两边呈现一定的夹角。这种结构严重影响了大π键的共轭,继而影响吸收光谱。罗丹明123的HOMO和LUMO轨道主要集中在氧杂蒽环上,苯酸酐对这两个轨道几乎没有贡献。因此罗丹明分子的吸收光谱和派洛宁分子的吸收光谱在一定程度上存在相似性。与派洛宁分子的吸收波长相比,罗丹明衍生物的吸收波长要小一些,原因是苯甲酸对氧杂蒽环上共轭效应的分裂作用影响。如果氧杂蒽环及其取代基相同,苯甲酸酐的存在大约能使吸收光谱蓝移30nm[11]。
比较三种派洛宁的吸收波长发现,派洛宁B的吸收波长大于派洛宁Y的吸收波长,大于派洛宁20的吸收波长。这是因为派洛宁B的氨基N(C2H5)2的供电子能力大于派洛宁Y的氨基N(CH3)2的共电子能力,大于派洛宁20的氨基NH(C2H5)的供电子能力。三种派洛宁衍生物主要的电子跃迁都是电子从HOMO到LUMO的跃迁,主要的前线轨道如图2所示。氨基对HOMO和LUMO都有一定的贡献,而且对LUMO的贡献要比HOMO的贡献要小,这也说明氨基对吸收光谱的影响。氧杂蒽环上的氧原子对LUMO都有一定的贡献。由图2所示,HOMO是典型的π轨道,LUMO是一个典型的π*轨道。因此三种派洛宁衍生物的吸收光谱主要是π→π*跃迁。


图2 派洛宁B 和罗丹明123的前线分子轨道。
3 结论
本文中使用量子化学计算方法在B3LYP/6-31G(d,p)理论水平上研究了氧杂蒽衍生物的吸收光谱。派洛宁衍生物的吸收光谱主要是π→π*跃迁。氨基的供电站能力越强,吸收波长越长。与派洛宁分子的吸收波长相比,罗丹明123的吸收波长要小一些,原因是苯甲酸对氧杂蒽环上共轭效应的分裂作用影响。
猜你喜欢
无机化学学报(2022年9期)2022-09-16
安徽化工(2022年2期)2022-04-12
沈阳大学学报(自然科学版)(2018年5期)2018-11-07
中国资源综合利用(2017年1期)2018-01-22
分析化学(2017年12期)2017-12-25
山东工业技术(2016年15期)2016-12-01
中国塑料(2016年1期)2016-05-17
读写算·教研版(2016年8期)2016-05-07
中国塑料(2016年11期)2016-04-16
中国粮油学报(2016年5期)2016-01-23
