
Taq DNA聚合酶5′~3′外切活性对荧光定量PCR的影响
2020-03-27 07:13:28蒋析文刘霭珊陈巍
分子诊断与治疗杂志 2020年2期
蒋析文 刘霭珊 陈巍
Taqman 法荧光定量PCR 是利用Taq DNA聚合酶(后简称Taq 酶)的5′~3′DNA 外切活性,在PCR 过程中切割与模板链结合的荧光探针,释放出荧光基团而产生信号[1]。在反应过程中,Taq DNA聚合酶的聚合酶活性区与5′~3′DNA 外切活性区均参与反应[2-4]。目前Taq 酶标定的浓度为聚合酶活性单位浓度[5-6],缺乏对5′~3′DNA 外切活性的定量。由于Taqman 探针法荧光定量PCR 信号的产生与Taq 酶的5′~3′DNA 外切活性直接相关,因此本文研究了不同热启动Taq 聚合酶的5′~3′DNA 外切酶活性差异,以及该活性对Taqman 探针法荧光定量PCR的影响,以期进一步了解Taq 酶两种不同活性区在反应中所起的作用,并指导Taqman 法荧光定量PCR 反应的优化。
1 方法
1.1 材料
Taq 酶、Hotstar Taq 酶、HotStar-II Taq 酶均为达安基因制备的原料产品,浓度均为10 U/μL。5′~3′DNA 外切酶活性检测底物为:Sub1:ATGCTGGTTAAGCTGCCGTAGTCATGCAGTACGTCCGT ACGTCA,Probe1:FAM-CAGCTTAACCAGCATBQH1。外切活性反应buffer(10X):500 mM Tris、500 mM KCl、20 mM MgCl2、2.5 mM dNTP、pH8.8。乙型肝炎病毒(hepatitis B virus,HBV)DNA 荧光定量PCR 检测试剂盒为达安基因产品。
1.2 方法
1.2.1 5′~3′DNA 外切酶活性的测定
酶样品用保存液进行梯度稀释:5、4、3、2、1、0 U/μL,反应体系见表1。

表1 5′~3′DNA 外切酶活性测定反应体系Table 1 The Reaction of 5′~3′exonuclease activity assay
反应体系放置于Q5 荧光定量PCR 仪中,设置荧光检测通道为FAM,设置反应程序:95℃10 min,(95℃10 s,55℃30 s 并读取荧光)X 40 个循环。反应后将原始荧光数据导出,以荧光强度(relative fluorescence units,RFU)为纵坐标,反应时间(min)为横坐标作图,得到在各浓度下的外切酶反应动力学曲线。计算反应前期的RFU 值线性增长部分(线性R2>0.9)的斜率,即为各聚合酶浓度梯度下外切酶活性测试的反应速率(RFU/min)。
1.2.2 不同酶的5′~3′DNA 外切酶活性的对比
根据1.2 测定的同一个聚合酶在不同聚合酶活性浓度下的外切酶活性反应速率,以反应速率为纵坐标,聚合酶活性浓度(U/μL)为横坐标作图,得线性关系方程,方程的斜率即为在单位聚合酶活性浓度下所具有的外切酶活性。对比不同酶所作线性关系方程的斜率,即可计算其外切酶活性的差异倍数。
1.2.3 不同酶在HBV 病毒DNA 荧光定量PCR 反应体系中的性能测试
取达安HBV 病毒DNA 荧光定量PCR 检测试剂盒,按照使用说明书的方法进行阳性参照品的提取制备。将酶样品稀释至1 U/μL,然后按使用说明每反应加入27 μL 反应液A 及3 μL 的1 U/μL酶样品,混合均匀后加入20 μL 阳性参照样品提取物。按95℃10 min,(95℃10 s,55℃30 s 并读取荧光)×40 个循环,进行PCR 反应。
2 结果
2.1 5′~3′DNA 外切酶活性的测定
5′~3′ DNA 外切酶活性的测定原理如图1。Taq 聚合酶的5′~3′DNA 外切酶活性不依赖于上游的模板及引物,对交叉DNA 或双链DNA 的5′游离末端具有特异性因此设计与单链DNA(Sub1)互补的探针(Probe1),探针5′端标记FAM 荧光,3′端标记BQH1 淬灭基团,当Taq 聚合酶5′~3′DNA 外切活性降解Probe1 时,FAM 荧光基团从探针脱离,发出荧光信号,通过实时荧光定量PCR 仪可检测出反应曲线(图2)。从图2 可见,随着聚合酶浓度的下降,反应曲线的斜率随之下降,反应曲线前期荧光线性增长区的斜率即为各浓度梯度下反应速率。从图3 可见,反应的初始速率与加入的聚合酶浓度成线性关系(r2=0.982),此直线的斜率可衡量单位浓度的Taq 酶所具有的5′~3′DNA 外切活性高低。
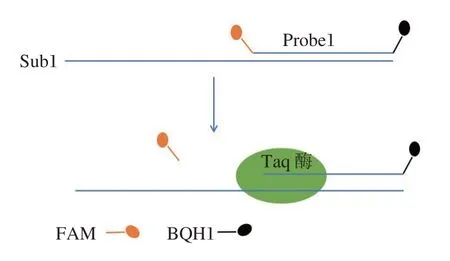
图1 5′~3′DNA 外切酶活性测定原理Figure 1 Schematic of 5′~3′exonuclease activity assay
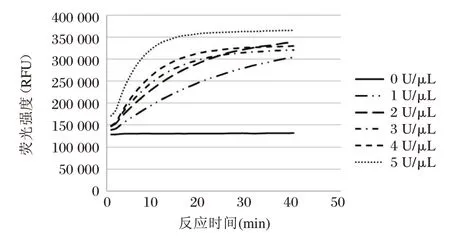
图2 不同浓度的Taq 酶与底物反应后的荧光曲线Figure 2 Fluorescence curve of substrate reaction with gradient of Taq
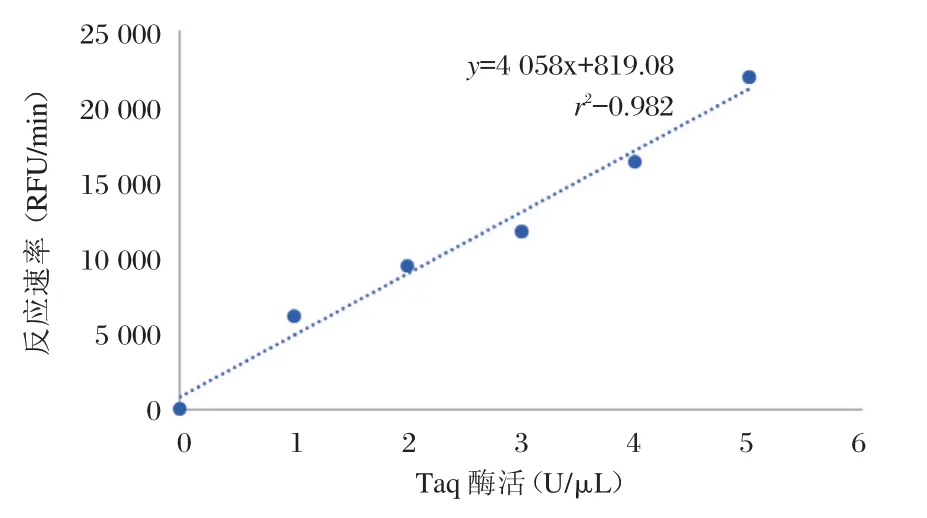
图3 Taq 酶浓度与其5′~3′DNA 外切活性的线性关系Figure 3 Linear relationship between the concentration of Taq and its 5′~3′exonuclease
2.1 不同热启动Taq 酶的5′~3′DNA 外切活性的对比
两种热启动酶Hotstar Taq 和Hotstar-II Taq 来源于Taq 酶,经不同化学修饰方法制备而成。测定3 种酶在相同浓度下的5′~3′DNA 外切活性,结果见图4。
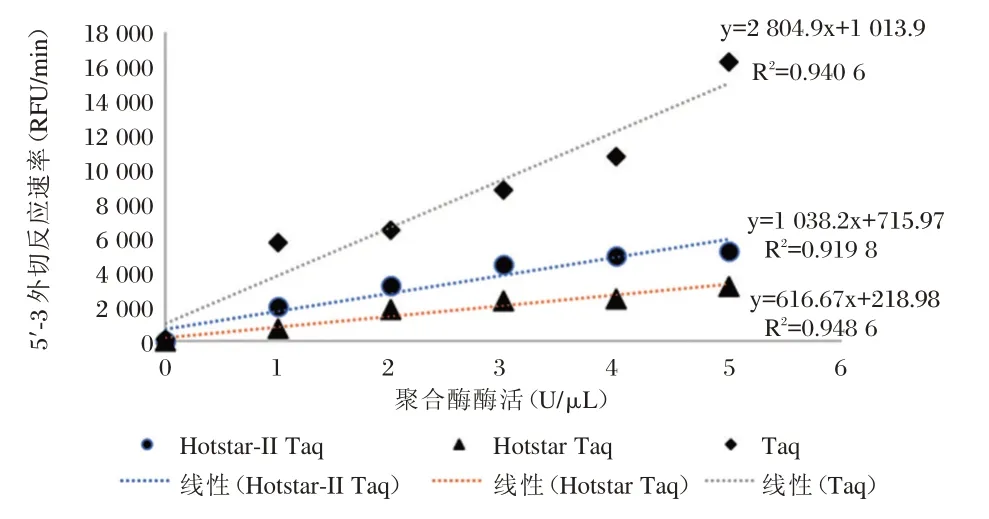
图4 Hotstar Taq、Hotstar-II Taq、Taq 酶在相同聚合酶活性浓度下的5′~3′外切活性的对比Figure 4 comparison of 5~3′exonuclease activity of Hotstar Taq、Hotstar-II Taq、Taq with the same polymerase activity
从图4 可见,未经化学修饰的Taq 酶同等聚合酶活性浓度下具有最高5′~3′外切活性,而经过化学修饰的制备而成的Hotstar Taq 及Hotstar-II Taq的5′~3′外切活性均有所下降,其中Hoststar Taq 5′~3′外切活性最低。
2.3 热启动酶5′~3′ DNA 外切活性对Taqman 探针法荧光定量PCR的影响
使用不同浓度的Hotstar Taq、Hotstar-II Taq 及Taq 酶,对4 个梯度的HBV 阳性参照物(2e6~2e3)进行荧光定量PCR 扩增。结果见图5。
从图5 可见,当每反应中加入3 U HotStar Taq、HotStar-II Taq、Taq 酶时,各阳性参照物梯度扩增的情况基本一致,荧光曲线重合;每反应中加入0.38 U、0.19 U 酶时,Taq 酶对各阳性参照物梯度扩增每循环产生的荧光强于HotStar-II Taq,Hot-Star-II Taq 强于HotStar Taq。证明酶对探针的外切反应为PCR 反应的限速步骤。
根据图4 中测定的各不同Taq 酶单位浓度下的5′~3′ DNA 外切活性(标准曲线方程的斜率),Taq 酶的外切活性为HotStar-II Taq 的2.70 倍,Hot-Star Taq 的4.55 倍,调整PCR 反应中酶的用量至反应中加入的5′~3′外切酶活性单位一致(Taq:0.38 U/反应,HotSatr Taq:1.73 U/反应,HotStar-II Taq 1.03 U/反应),测试对HBV 阳性参照物样品的扩增效率,结果见图6。从图6 可见,即使各酶加入量不同,但扩增曲线基本一致,证明当反应中5′~3′外切酶活性一致时,增加聚合酶活性并没能进一步提高PCR的扩增效率,因此进一步证明了酶对探针的外切反应为PCR 反应的限速步骤。

图5 3 种聚合酶活性不同活性梯度下对HBV 阳性参照物的测试结果Figure 5 Amplification of HBV positive sample by three polymerases with equal activity unit

图6 3 种聚合酶扩增不同梯度HBV 阳性参照物的测试结果Figure 6 Amplification of gradient HBV positive sample by three polymerases with different activity unit
3 讨论
Taqman 探针法实时荧光定量PCR 是当今分子诊断的主流技术,以其快速、高通量[7]、多通道[8]、高灵敏度[1,9]、高特异性[2]等特点,被广泛应用于病原微生物的定性及定量检测[10-12]、单核苷酸多 态(Single nucleotide polymorphism,SNP)检测[13-14]、短串联重复序列(Short Tandem Repeat,STR)分型检测[15]、基因表达水平差异检测[16]、等位基因表达差异检测(Allelic Expression Imbalance,AEI)[17]、融合基因检测[4]等方面,在科研、临床诊断、疾病防控、兽医、法医、检验检疫等领域有着广阔的应用前景。其检测原理是:具有5′~3′DNA 外切活性的DNA聚合酶在PCR 延伸过程中遇到结合在模板链上的荧光标记探针时,利用其5′~3′DNA 外切活性将探针降解,释放荧光信号并由实时定量PCR 仪检测[5]。根据此原理,Taqman探针法荧光定量PCR 检测的信号与PCR 产物的扩增及探针的切割直接相关。在Taq DNA聚合酶上,聚合酶活性区与5′~3′DNA 外切活性区位于不同的结构域[15],聚合活性区与外切活性区共同作用,驱动新生DNA 链合成的同时切割荧光探针释放信号。在聚合与外切两种不同的反应中,反应效率低的一方直接决定DNA 扩增的效率以及荧光信号释放的效率。根据报道,Taq 酶并非完全降解探针,其降解的区域从探针的5′端起约5~12 bp,未完全降解的探针有可能参与后续的PCR 循环,从而抑制荧光信号的释放,而探针的降解与Taq 酶的外切活性的强弱密切相关[3,18]。从本研究所得结果可见,当反应中酶过量时(3 U/反应),聚合酶活性及外切酶活性同时满足反应的需求,不同酶之间扩增效率没有明显差异。但在酶量不足的情况下(0.38、0.19 U/反应),反应中加入相同聚合酶活性的Taq 酶,具有更高外切酶活性的酶其扩增效率更高;而加入相同外切酶活性的Taq 酶,即使聚合酶活性不同,但扩增效率基本一致。以上结果证明外切反应为限速步骤,其速率决定DNA扩增的效率。
提高Taq 酶的DNA 聚合活性及持续延伸能力是改进PCR 效率的一贯策略,但由于Taqman 探针法荧光定量PCR的信号产生依赖于探针的降解,而本研究揭示了Taq 酶5′~3′ DNA 外切活性与PCR 荧光信号的关系,为Taq 酶的性能改造指出了新的方向。为提高PCR 特异性,均需要对Taq酶的活性位点进行封闭修饰从而达到热启动的目的。化学修饰由于成本较低,能在较高温度下保持封闭,是对Taq 酶进行热启动修饰的较常用方法。但化学修饰不可避免地对Taq 酶的结构和活性有影响,在修饰过程中会出现蛋白变性、沉淀和失活,被修饰基团在酶上各结构域分布的情况也可能影响各结构域在修饰后性能改变的程度[19]。本研究中两种不同化学修饰制备的Taq 酶表现出不同的外切酶活性和PCR 扩增性能也证明了这一弊端。抗体修饰Taq 酶或配体修饰Taq 酶由于通过与Taq酶结合而屏蔽活性位点达到热启动的目的[20-21],因此能保留Taq 酶完整的活性。在耐热DNA聚合酶的突变改造中,突变筛选方法通常专注于提高聚合酶性能而忽视对外切酶性能的考察[6,22-23],而抑制或删除5′~3′外切活性结构域已被证明是提高聚合酶性能的有效途径[25],因此筛选出的突变体很可能因外切酶活性低而不适用于Taqman 探针法定量PCR。为解决这一问题,有必要在筛选前确认突变体具有完整的外切活性功能。
猜你喜欢
中等数学(2022年1期)2022-06-05 07:50:08
科技创新与应用(2018年23期)2018-09-13 10:47:16
中等数学(2018年4期)2018-08-01 06:36:34
中学数学研究(广东)(2018年23期)2018-03-05 07:54:32
吉林省教育学院学报(2017年12期)2018-01-29 18:48:54
初中生世界·九年级(2017年9期)2017-10-13 10:27:04
现代检验医学杂志(2016年3期)2016-11-15 01:59:28
三峡大学学报(自然科学版)(2016年6期)2016-04-16 05:02:56
当代化工研究(2016年7期)2016-03-20 16:22:02
物理实验(2015年9期)2015-02-28 17:36:47
