
分散微固相萃取-高效液相色谱-串联质谱法测定水样中3种氯霉素类和11种激素类药物残留
2020-03-21 01:18粟有志李艳美王兴磊
分析科学学报 2020年1期
粟有志 李艳美 周 均 尚 爽 李 芳 王兴磊
(1.伊犁师范大学污染物化学与环境治理重点实验室新疆伊宁 835000;2.中华人民共和国伊宁海关新疆伊宁 835000)
氯霉素类和激素类药物是临床治疗、畜牧水产、个人护理等领域常用药品。目前由于这些药品的滥用,两类药物已成为扰乱人体及动物正常生命活动的环境污染物[1]。近年来,水环境中氯霉素类和激素类药物残留常有检出,浓度在ng/L~μg/L范围[2,3]。氯霉素类和激素类残留的分析方法主要有液相色谱法[4-6]、液相色谱-质谱联用法[7-9]、气相色谱-质谱联用法[10,11]。由于水环境中此两类药物的含量很低,常需有效的前处理技术进行富集和净化,其中液相微萃取[7,11]和固相萃取(SPE)[8-10]是主要的前处理技术。分散微固相萃取技术(DMSPE)原理是将待净化提取液直接加入含有固体填料的离心管中,通过涡旋、振荡、离心等方式将待测目标物吸附于填料上,而后通过解吸溶剂进行洗脱。DMSPE相较于传统的SPE技术,无需繁琐的上样、淋洗、平衡等过程,具有消耗溶剂少、价格低廉、耗时短等特点,已在水环境、牛奶等样品的药物残留分析中得到广泛应用[12,13]。
本研究以官能化聚苯乙烯/二乙烯苯填料(PEP)作为吸附净化材料,采用DMSPE技术对河水和自来水进行前处理,结合高效液相色谱-串联质谱法(HPLC-MS/MS),建立了快速测定水样中3种氯霉素类:甲砜霉素(Thiamphenicol,TPC)、氟苯尼考(Flurbenicol,FBC)、氯霉素(Chloramphenicol,CRP),以及11种激素类:诺龙(Nandrolone,NL)、己烯雌酚(Diethylstilbestrol,DTSB)、己二烯雌酚(Dienetrol,DNT)、睾酮(Testosterone,TTT)、己烷雌酚(Hexoestrol,HXT)、甲基睾酮(Methyltestosterone,MTT)、甲地孕酮(Megestrol,MGT)、安宫黄体酮(Medroxyprogesterone Acetate,MPA)、美仑孕酮(Melengestrol,MLT)、孕烯二酮(Progesterone,PGT)、丙酸睾酮(Testosterone propionate,TTP)14种药物残留的分析方法。
1 实验部分
1.1 仪器与试剂
1200型高效液相色谱仪(Agilent公司);API5000型三重四极杆质谱仪(Applied Biosystems公司);3-18K型台式冷冻离心机(Sigma公司);N-EVAP-112型氮吹仪(Organomation公司);MS3型涡旋振荡器(IKA公司)。
14种药物标准品均购自Dr.Ehrenstorfer公司。分别准确称取各标准品10 mg(精确至0.0001 g)于10 mL容量瓶中,用甲醇溶解并定容至刻度,配成质量浓度为1 000 mg/L的单一标准储备液,于冰箱4 ℃储存备用。以乙腈-水(1∶9,V/V)为稀释液,配制10 mg /mL的中间混合标准溶液。乙腈、甲醇、甲酸均为色谱纯(Merck公司);酸性氧化铝(ALA)、N-丙基乙二胺(PSA)(40 μm)吸附填料购自Agilent公司;官能化聚苯乙烯/二乙烯苯极性增强聚合物(PEP,80~10 μm)、强阴离子交换混合机理的水可浸润型聚合物(PAX,40~60 μm)、强阳离子交换混合机理的水可浸润型聚合物(PCX,40~60 μm)、弱阳离子交换混合机理的水可浸润型聚合物(PWCX,40~60 μm)、强阴离子交换机理的硅胶(SAX,50 μm)、新型碳黑材料(PestiCarb,120~400 mesh)、官能化聚苯乙烯/二乙烯苯中等极性聚合物(HXN,40~60 μm)等吸附填料均购自Agela公司。实验用水由Milli-Q Advangtage A10超纯水系统(Millipore公司)制备。
1.2 样品采集与保存
河水采自伊犁河伊宁市段,自来水采自实验室。样品采集于棕色玻璃瓶中,采集水样前,先用水样洗涤取样瓶及盖子2~3次,充满至溢出后盖上盖子,于4 ℃保存,保存时间一般不超过7 d。
1.3 样品前处理
称取50±5 mg PEP吸附填料,置于50 mL离心管中,加入2 mL甲醇,先以3 000 r/min涡旋30 s,再以8 000 r/min离心1 min,弃去上层甲醇,在离心管中准确加入50 mL水样,以3 000 r/min涡旋30 s,再以8 000 r/min离心1 min,移出上层水样至离心管中(剩余约4 mL水样)。将剩余上层水样全部转移至装有0.45 μm尼龙膜的5 mL注射器中,过滤,弃去滤液。在离心管中加入4 mL甲酸-甲醇(1∶99,V/V),然后以3 000 r/min涡旋30 s,8 000 r/min离心1 min,将上层甲酸-甲醇转移至原来装有0.45 μm尼龙膜的5 mL注射器中,过滤,收集滤液于15 mL玻璃试管中。加入4 mL甲酸-甲醇重复操作一次,合并两次洗脱液,于40 ℃水浴中氮吹至干,用0.5 mL乙腈-水(1∶9,V/V)溶解定容后,供HPLC-MS/MS分析。
1.4 LC-MS/MS条件
色谱条件:JADE-PAK CB-C18柱(100 mm×2.1 mm,3 μm);流动相:A相为水溶液,B相为甲醇。梯度洗脱程序如下:0~0.5 min(B相10%),0.5~1.0 min(B相10%~70%),1.0~6.0 min(B相70%~95%),6.0~7.0 min(B相95%),7.0~7.5 min(B相95%~10%),7.5~12.0 min(B相10%)。柱温:30 ℃;流速:0.25 mL/min,进样量:10 μL。
质谱条件:电喷雾电离源(ESI),正、负离子扫描,以多重反应监测(MRM)模式检测;实验所用的气体均为高纯氮;碰撞气(CAD)压力为41 kPa;气帘气(CUR)压力为150 kPa;雾化气(GS1)压力为450 kPa;辅助气(GS2)压力为410 kPa;去溶剂温度500 ℃;ESI-模式下,电喷雾电压(IS)为-4.5 kV,预四极杆Q0入口电压(EP)为-10 V,碰撞室出口电压(CXP)为-22 V;ESI+模式下,IS为5.5 kV,Q0 EP为10 V,CXP为22 V。14种药物的定性/定量离子对、碰撞能量(CE)及去簇电压(DP)见表1。
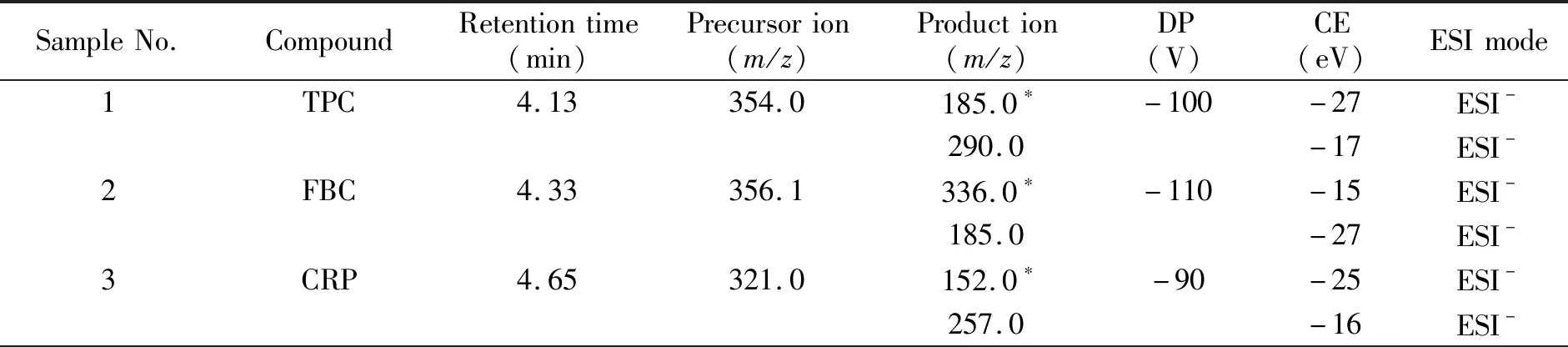
表1 3种氯霉素类和11种激素类药物的质谱分析参数Table 1 Optimized parameters of MS/MS for 3 Chloramphenicols and 11 Hormones
(续表1)
*quantification transition.
2 结果与讨论
2.1 仪器条件的优化
配制50 ng/L的单一药物的标准溶液,分别采用ESI+和ESI-进行母离子扫描,确定其准分子离子峰。选择信号强的分子离子峰进一步对子离子、去簇电压、碰撞能量等参数进行优化,参照欧盟指令(657/2002/EEC)决议中有关规定,本文选择离子丰度最高、本底干扰最小的两个离子对作为特征离子对,其中信号较大的离子对作为定量离子。实验比较了不同浓度的甲酸水溶液、乙酸铵水溶液、甲酸和乙酸铵的水溶液、纯水所组成的无机相(A相),乙腈、甲醇组成的有机相(B相)。通过不同A相与B相的组合分析发现:适合于ESI+的药物以甲酸水溶液-乙腈为流动相时响应信号较高;适合于ESI-的药物以水-甲醇为流动相时响应信号较高。综合考虑正负离子的响应信号,本研究最终选择水-甲醇为流动相。通过优化梯度洗脱程序,14种药物均能得到较好的分离度和色谱峰形。基于优化结果,建立的多重反应监测(MRM)质谱分析参数见表1。
2.2 DMSPE条件的优化
2.2.1 吸附剂和洗脱剂的选择比较了ALA、PSA、PEP、PAX、PCX、PWCX、SAX、PestiCarb和HXN 9种不同类型吸附剂对3种氯霉素类和11种激素类药物的吸附效果。移取50 mL空白河水,加入中间混合标准溶液至各药物的浓度均为50 ng/mL,分别加入0.1 g吸附剂,涡旋30 s进行吸附,计算吸附剂对14种药物的吸附率。结果表明:ALA的吸附率为19.5%~81.5%,PSA的吸附率为13.0%~82.1%,SAX的吸附率为9.7%~81.3%,其余6种吸附剂的吸附率均在98%以上。分别以10 mL乙腈、甲醇、甲酸-甲醇(1∶99,V/V)、氨水-甲醇(5∶95,V/V)为洗脱剂进行解吸实验。结果发现,甲醇和乙腈的洗脱率普遍低于甲酸-甲醇(1∶99,V/V)和氨水-甲醇(5∶95,V/V)的洗脱率。以甲酸-甲醇洗脱时,PestiCarb的洗脱率为1.4%~50.0%,HXN的洗脱率为42.1%~99.5%,PAX的洗脱率为50.5%~111.2%,PWCX的洗脱率为58.7%~101.3%,PCX的洗脱率为54.1%~93.6%,PEP的洗脱率为81.6%~110.8%。以氨水-甲醇为洗脱剂时,PestiCarb的洗脱率为2.2%~25.5%,HXN的洗脱率为42.1%~98.2%,PAX的洗脱率为60.4%~108.2%,PWCX的洗脱率为52.9%~92.2%,PCX的洗脱率为58.7%~95.5%,PEP的洗脱率为66.8%~116.6%。因PestiCarb对目标物的吸附容量过大,难以将目标物解吸,洗脱率偏低;PAX、PWCX、PCX 3种吸附剂具有聚合物均衡吸附和离子交换吸附的双重机理,洗脱剂对部分目标物的洗脱率较差;HXN难以满足NL、DNT、TTT 3种药物的洗脱率同时满足方法学要求。实验以PEP为吸附剂,以甲酸-甲醇(1∶99,V/V)为洗脱剂,目标物的回收率较好。
2.2.2 吸附剂用量的选择移取50 mL空白河水,加入中间混合标准溶液至各药物的浓度均为50 ng/mL。分别称取25、50、100、150、200、300 mg PEP吸附剂进行吸附实验。结果表明:用量25 mg时,除己二烯雌(DNT)的吸附率为94.8%,其他不同用量条件下,14种药物的吸附率均在98%以上。本研究选择吸附剂PEP的用量为50 mg。
2.2.3 吸附时间的选择称取50 mg PEP吸附剂,加入50 mL各药物浓度为50 ng/mL的河水,比较涡旋15、30、60、90、120 s不同涡旋时间对吸附效率的影响。结果发现,在上述范围内,14种药物的吸附效率均在98%以上。考虑到15 s时间过短,可能影响吸附效果,本研究最终选择涡旋30 s。
2.2.4 水样pH的选择移取50 mL空白河水,加入中间混合标准溶液至各药物的浓度均为50 ng/mL,用稀HCl或NaOH溶液,分别将河水pH调至3、5.8、8.5进行吸附实验。结果表明,3种pH条件下,14种药物的吸附率均在98%以上。因所采集水样pH在6.7~7.6范围,故实验无需调节pH。
2.2.5 洗脱剂的体积的选择在50 mL空白河水中加标至各药物的浓度为0.5 ng/mL,吸附完成后,用16 mL甲酸-甲醇(1∶99,V/V)分4次洗脱(每次用量4 mL),分别收集每次洗脱液氮吹至干,用0.5 mL乙腈-水( 1∶9,V/V)定容后分析。结果表明,当进行第3次洗脱时,回收率无明显提高。故选择8 mL甲酸-甲醇分2次洗脱。经DMSPE前处理的河水加标样品的MRM色谱图见图1。
2.2.6 与常规SPE方法的对比对比固相萃取小柱Agela Cleanrt PEP(60 mg/3mL)、Waters Oasis HLB(60 mg/3mL)和DMSPE方法对3种氯霉素类和11种激素类药物的实验回收率。PEP和HLB固相萃取预先用3 mL甲醇和3 mL水活化,上样50 mL加标浓度为500 ng/L的河水样品,用3 mL水淋洗,抽干,用8 mL甲酸-甲醇(1∶99,V/V)洗脱,收集洗脱液,40 ℃水浴中氮气吹至干,用0.5 mL乙腈-水(1∶9,V/V)定容溶解。3种不同处理方法对14种目标分析物的回收率无明显差别(图2)。
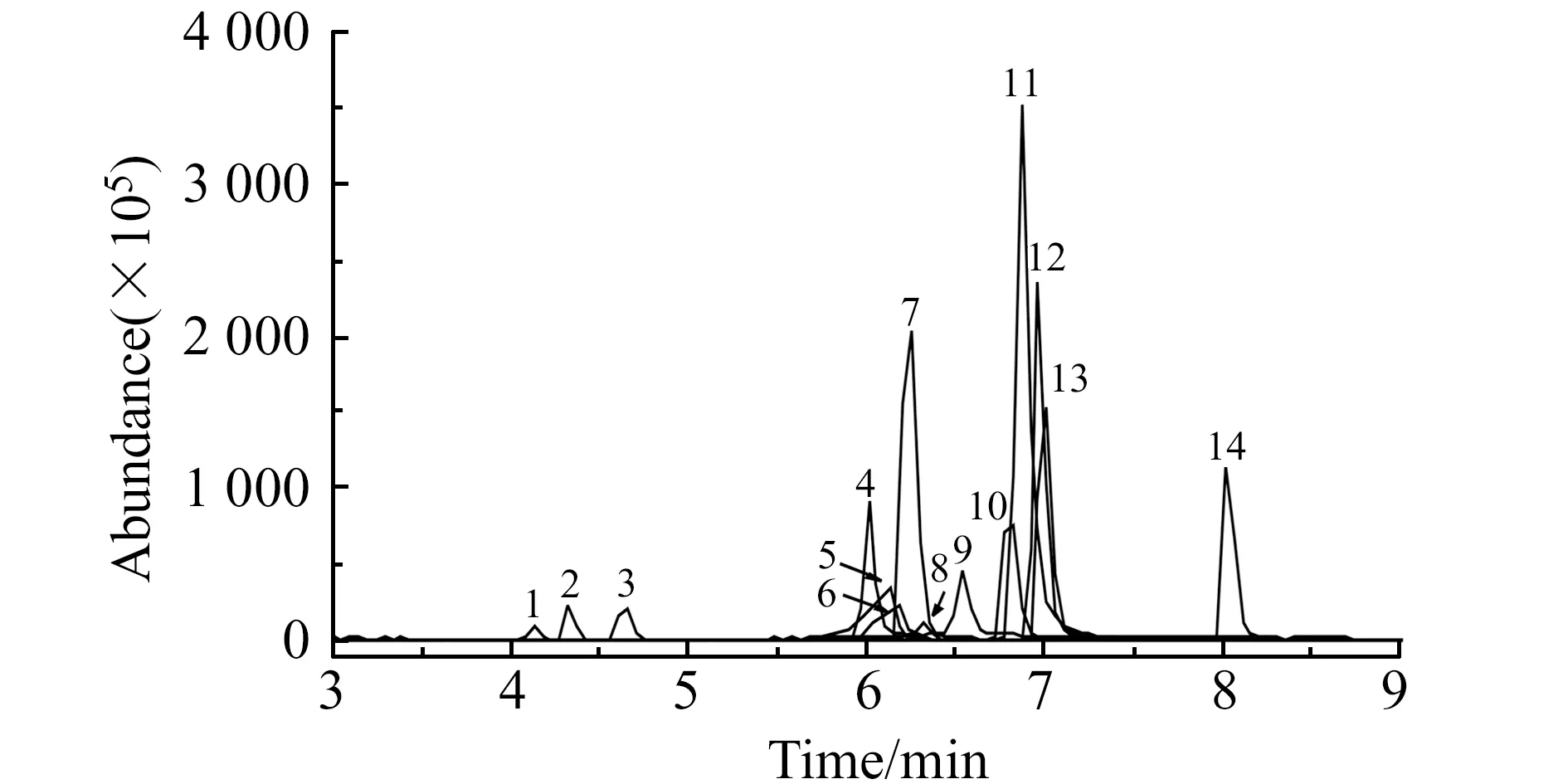
图1 空白河水中添加浓度为10 ng/L的3种氯霉素类和11种激素类药物的MRM色谱图Fig.1 MRM chromatograms of 3 chloramphenicols and 11 hormones adding concentration of 10 ng/L in blank river waterNo.1-14 are the same as Table 1.
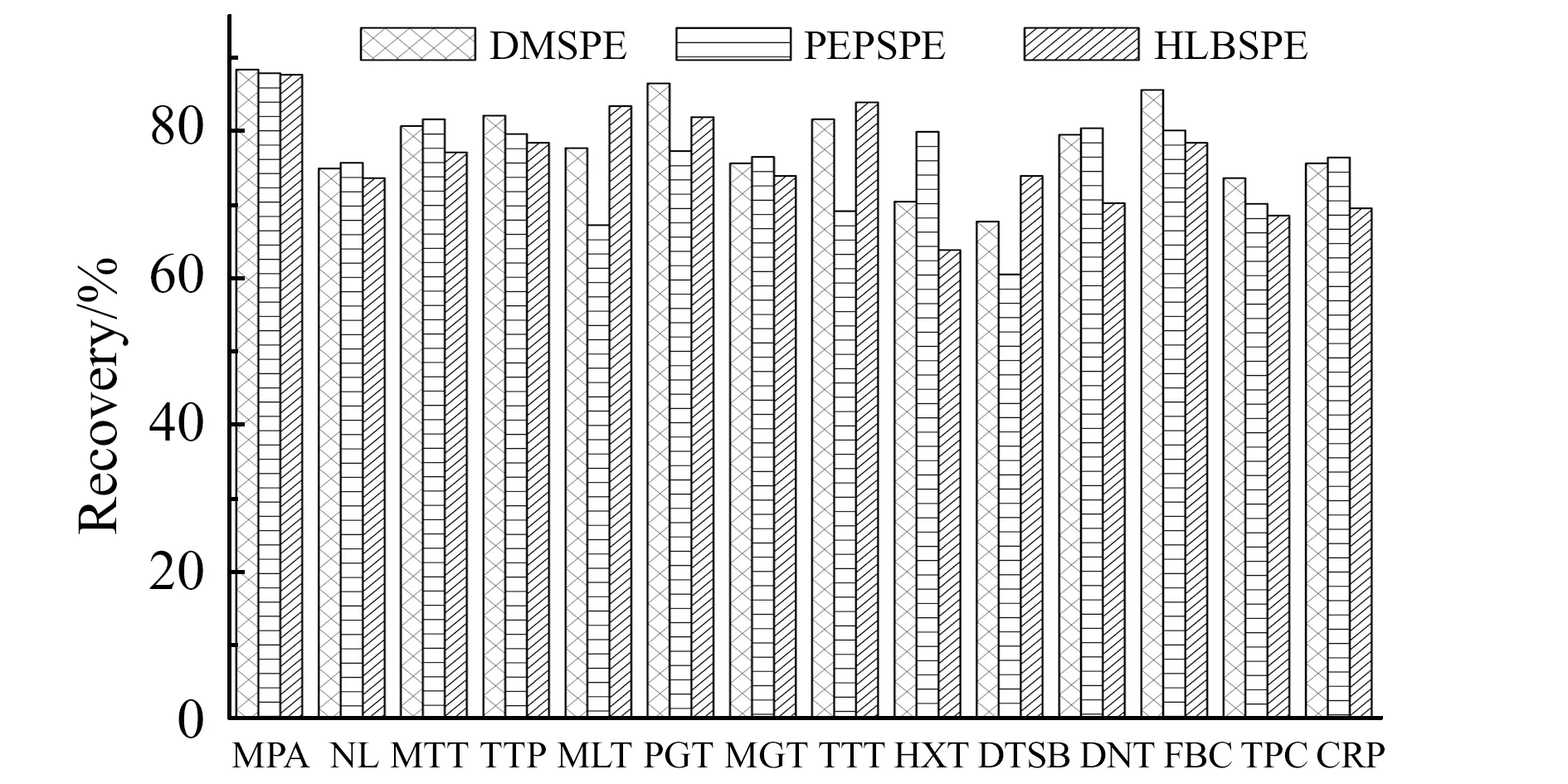
图2 三种不同前处理对14种目标分析物的回收率Fig.2 The recovery of 14 target analytes by three different pretreatments
2.3 基质效应、线性范围、检出限和定量限
采用相对响应值法根据公式计算基质效应(ME):ME=Am/Ac×100%。其中Am表示空白基质标准响应值;Ac表示纯溶剂标准响应值。基质效应为87.5%~106.5%,均可忽略。将中间混合标准溶液逐级稀释成0.05、0.1、0.2、0.5、1、2、5、10、20、50 ng/mL的标准曲线工作溶液。以峰面积(Y)对分析物的质量浓度(X)作线性回归,14种药物的相关系数(r2)大于0.994。检出限按照信噪比(S/N)≥3确定,其范围为0.1~3.1 ng/L;以S/N≥10确定定量限,其范围为0.2~10.4 ng/L。14种药物的标准曲线线性范围、相关系数、基质效应、检出限与定量限详见表2。
2.4 方法的准确度和精密度
实验选择了河水、自来水的空白样品,设3个加标浓度,浓度分别为2、10、100 ng/L,每个水平重复6次。河水和自来水的回收率和相对标准偏差见表3。14种药物的回收率在53.6%~90.4%之间,日内精密度为5.2%~16.9%。
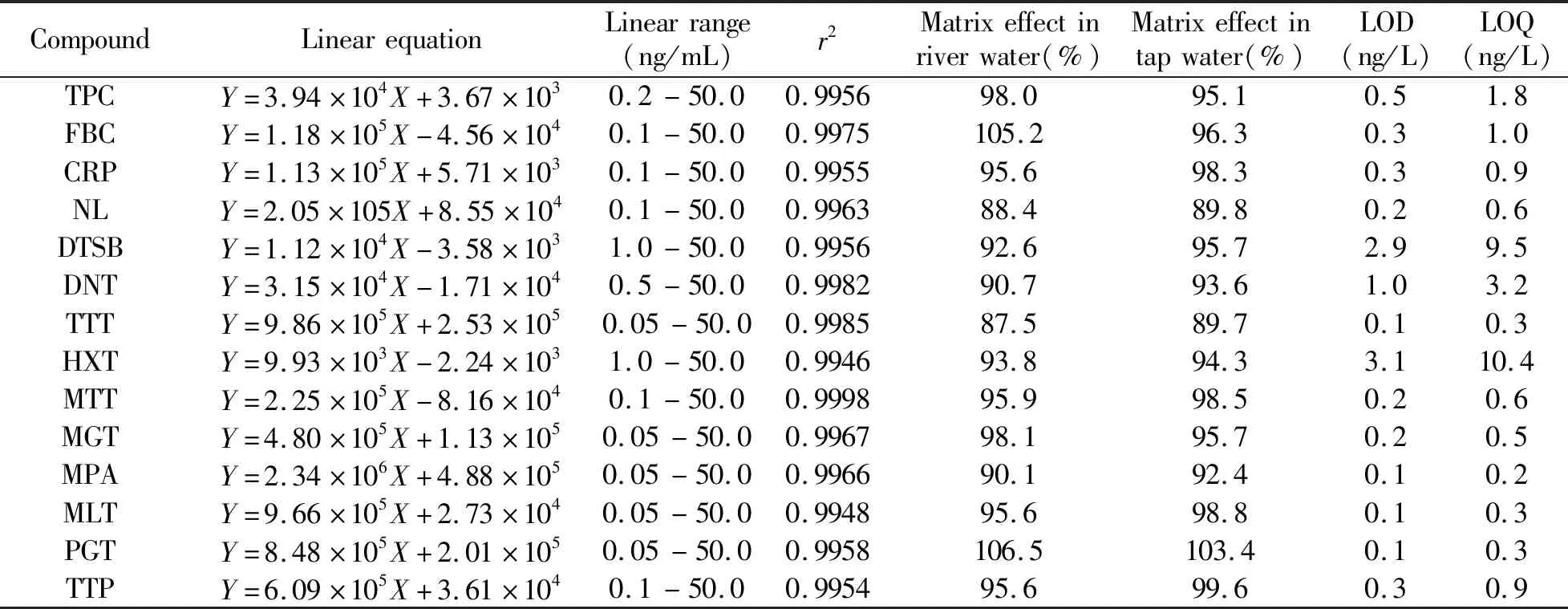
表2 3种氯霉素类和11种激素类药物的线性范围、标准曲线、相关系数、基质效应、检出限与定量限Table 2 Linear equation,linear ranges,correlation coefficients (r2),matrix effec,limits of determination (LOD) and limits of quantification (LOQ) of 3 chloramphenicol and 11 hormones
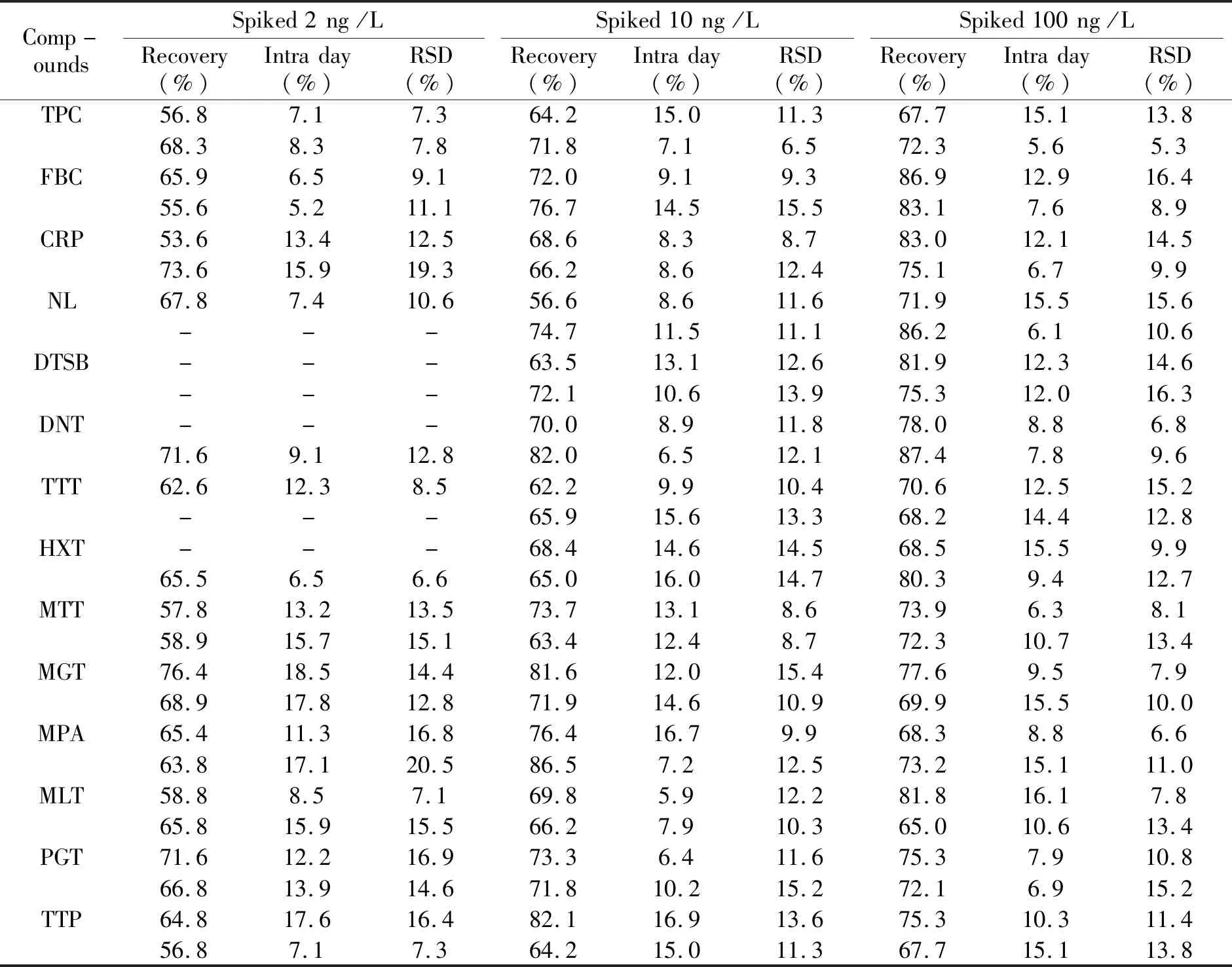
表3 3种氯霉素类和11种激素类药物的在河水和自来水中的添加回收率及相对标准偏差(n=6)Table 3 Recoveries and relative standard deviations (RSDs) of the 3 chloramphenicols and 11 hormones in river water(n=6)
2.5 样品测定
利用本方法对伊犁河伊宁市段的三个采样区域(城市上游、中游、下游)的河水及实验室自来水共计10个样品进行分析,均未检出3种氯霉素类和11种激素类药物。
3 结论
本研究建立了水样中14种药物残留的液相色谱-串联质谱分析方法。方法的定量限、回收率和精密度均满足痕量分析要求,可为水中此类物质的监控提供技术支持。
猜你喜欢
家庭医药(2022年5期)2022-05-18
核化学与放射化学(2022年2期)2022-04-28
食品安全导刊(2021年20期)2021-08-30
爱你(2018年8期)2018-11-14
钻井液与完井液(2018年2期)2018-06-13
爱你·健康读本(2018年3期)2018-05-14
爱你·健康读本(2018年3期)2018-05-14
中国蜂业(2018年4期)2018-05-09
家庭科学·新健康(2016年5期)2016-05-12
中南民族大学学报(自然科学版)(2015年2期)2015-12-16
