
Comparative analysis of mitochondrial genome of a deepsea crab Chaceon granulates reveals positive selection and novel genetic features*
2020-03-19 12:31ZHANGBoWUYingyingWANGXinJIANGWeiYINJianpingLINQiang
ZHANG Bo , WU Yingying , WANG Xin JIANG Wei , YIN Jianping LIN Qiang
1 Key Laboratory of Tropical Marine Bio-Resources and Ecology, South China Sea Institute of Oceanology, Chinese Academy of Sciences, Guangzhou 510275, China
2 Institution of South China Sea Ecology and Environmental Engineering, Chinese Academy of Sciences, Guangzhou 510275,China
3 University of Chinese Academy of Sciences, Beijing 100049, China
4 Beijing Advanced Sciences and Innovation Center of Chinese Academy of Sciences, Beijing 100049, China
5 Institute of Oceanology, Chinese Academy of Sciences, Qingdao 266071, China
Abstract Deep-sea organisms survive in an extremely harsh environment. There must be some genetic adaptation mechanisms for them. We systematically characterized and compared the complete mitochondrial genome (mitogenome) of a deep-sea crab ( Chaceon granulates) with those of shallow crabs. The mitogenome of the crab was 16 126 bp in length, and encoded 37 genes as most of a metazoan mitogenome, including 13 protein-coding genes (PCGs), 22 transfer RNA ( tRNA) genes, and 2 ribosomal RNA ( rRNA) genes. The gene arrangement and orientation was conserved in the crabs. However, a unique mitogenome element regulator,the origin of light-strand replication (O L), was f irstly predicted in the present crab mitogenome. In addition,further positive selection analysis showed that two residues (33S in ND3 and 502I in ND5) in C. granulates mitogenome were positively selected, indicated the selective evolution of the deep-sea crab. Therefore, the mitogenome of deep-sea C. granulates showed a unique O L element and positive selection. These special features would inf luence the mitochondrial energy metabolism, and be involved in the adaptation of deepsea environment, such as oxygen def icits and low temperatures.
Keyword: deep-sea organisms; mitochondrial genome; adaptation; Chaceon granulates
1 INTRODUCTION
The deep-sea is the most extensive ecosystem on earth (Rex, 1981). Unlike terrestrial and shallow organisms, deep-sea organisms survive in an extremely harsh environment, which including hundreds of bars of pressure, low oxygen, scarce food, constant darkness, and low temperatures(Sanders and Hessler, 1969). Because of the sparse animal life and technical diffi culties in sampling the deep-sea benthos, most knowledge regarding deepsea organisms is restricted to marine microbes and morphological distinctions of a few animal species.There is little information regarding the adaptive genetic mechanisms in the large deep-sea organisms(Etter et al., 1999; Sogin et al., 2006; Jebbar et al.,2015; Zhang et al., 2015; Coscia et al., 2018).
Mitochondria is one of the most essential organelle in eukaryotic cells (Bernt et al., 2013), and plays an important role in energy metabolism and various biosynthetic pathways (Green and Reed, 1998;Newmeyer and Ferguson-Miller, 2003). Mitochondrial DNA is more strongly inf luenced by evolutionary processes than nuclear DNA largely because it has a smaller eff ective population size and does not undergo recombination. The mitochondrial genome(mitogenome) has been widely used for study in genetic diversity, phylogeography, phylogenetic relationships, and adaptive mechanisms (Da et al.,2008; Hassanin et al., 2009; Yu et al., 2011; Jin et al.,2015; Liao et al., 2016). Recently, several mitochondrial genes have shown signif icantly adaptive evolution, including the cytochrome b gene of alpacas (Da et al., 2008), the cytochrome c oxidase gene of plateau pikas (Luo et al., 2008), the NADH dehydrogenase 6 gene of domestic horses (Ning et al.,2010), and the ATP synthase genes of Caprinae(Hassanin et al., 2009).
Crabs are typical benthic organisms and distribute all over the world. To adapt to diff erent environments,they have evolved a broad range of phenotypes. Thus,it can be used as an index for the study of adaptive mechanisms to some local environment. The mitogenomes of crabs are typically closed circular molecules, with 14 kb to 18 kb in length. They encode the following 37 genes: 13 protein-coding genes(PCGs), 22 transfer RNA (tNRA) genes, 2 ribosomal RNA (rRNA) genes, and a putative control region(CR) (Liu and Cui, 2010; Ma et al., 2013; Tang et al.,2017). However, the basic genetic information of crabs under diff erent environments is far from enough,especially for extremely environment. To date,Chaceongranulates(NC_AB769383) was sequenced,but the genomic characteristics have not been illuminated in detail. In this study, the complete mitogenome of another deep-seaC.granulateswas sequenced. Furthermore, in comparison with those of shallow water crabs, some unique genomic features were characterized. Interestingly, some novel genetic features that could be involved in the adaptation of deep-sea environment were identif ied. This work made up the data of crab genome, and should be useful for studies on crab evolution and adaptive mechanisms.
2 METERIAL AND METHOD
2.1 Specimens and DNA extraction
TheC.granulatesspecimen was collected by deep-water research vessel on December 15, 2014, on the Yap seamount (137.46° E; 8.52° N) in the Pacif ic Ocean at 477 m depth. The specimen was not a member of an endangered or protected species, and no specif ic permits were required. The specimen was stored in 99% ethanol and kept at 4°C. DNA was extracted using a Genomic DNA Kit (Tiangen Co.,Beijing, China) according to the manufacturer’s instructions.
2.2 PCR and sequencing
The complete mitogenome was amplif ied by using overlapping long-PCR. Five pairs of primers were designed at conserved regions of crab mitogenome(Supplementary Table S1). All PCR reactions were performed in 50 μL volume, with 1 μL of template DNA (approximately 100 ng), 0.3 μmol/L of each primer, 5 μL of 10 × LA Taq buff er (Mg2+plus), 5 μL of dNTP Mix (2.5 mmol/L), and 1 U of LA Taq(TaKaRa, Japan). The PCR amplif ications were performed according to the following procedure: one cycle of denaturation for 5 min at 94°C, 30 cycles of 40 s at 94°C, 40 s at the primer-specif ic annealing temperature, 5 min at 72°C, and f inally a 10-min extension at 72°C. After purif ication (TIANgel Midi Purif ication Kit DP209, Beijing), the PCR products were directly sequenced in both directions by using the PCR primers. Sequencing was performed by Thermo Fisher Scientif ic (Guangzhou, China).
2.3 Sequence assembly and annotation
The sequence alignments were conducted using Clustal X. The PCGs and rRNA genes were identif ied with BLAST and the NCBI database, and were compared with the mitogenome sequences of several other crab species (Supplementary Table S2). The tRNA genes and their secondary structures were predicted by using the web-based tRNAscan-SE 1.21(Lowe and Eddy, 1997). The tRNASer, which was not found by using the software tools, was identif ied on the basis of sequence similarity to the published crab mitochondrial tRNASer(Ma et al., 2015; Miller et al.,2005). The skew in the nucleotide composition was calculated with AT-skew and GC-skew and measured according to the following formulae: AT-skew=(A-T)/(A+T) and GC-skew=(G-C)/(G+C) (Perna and Kocher, 1995), where A, T, C, and G are the occurrences of the corresponding bases. The codon usage was calculated with the Codon Usage Database(http://www.kazusa.or.jp/codon/). The gene map of the complete mitogenome was illustrated using OGDRAW (http://ogdraw.mpimp-golm.mpg.de/).
2.4 Positive selection analysis
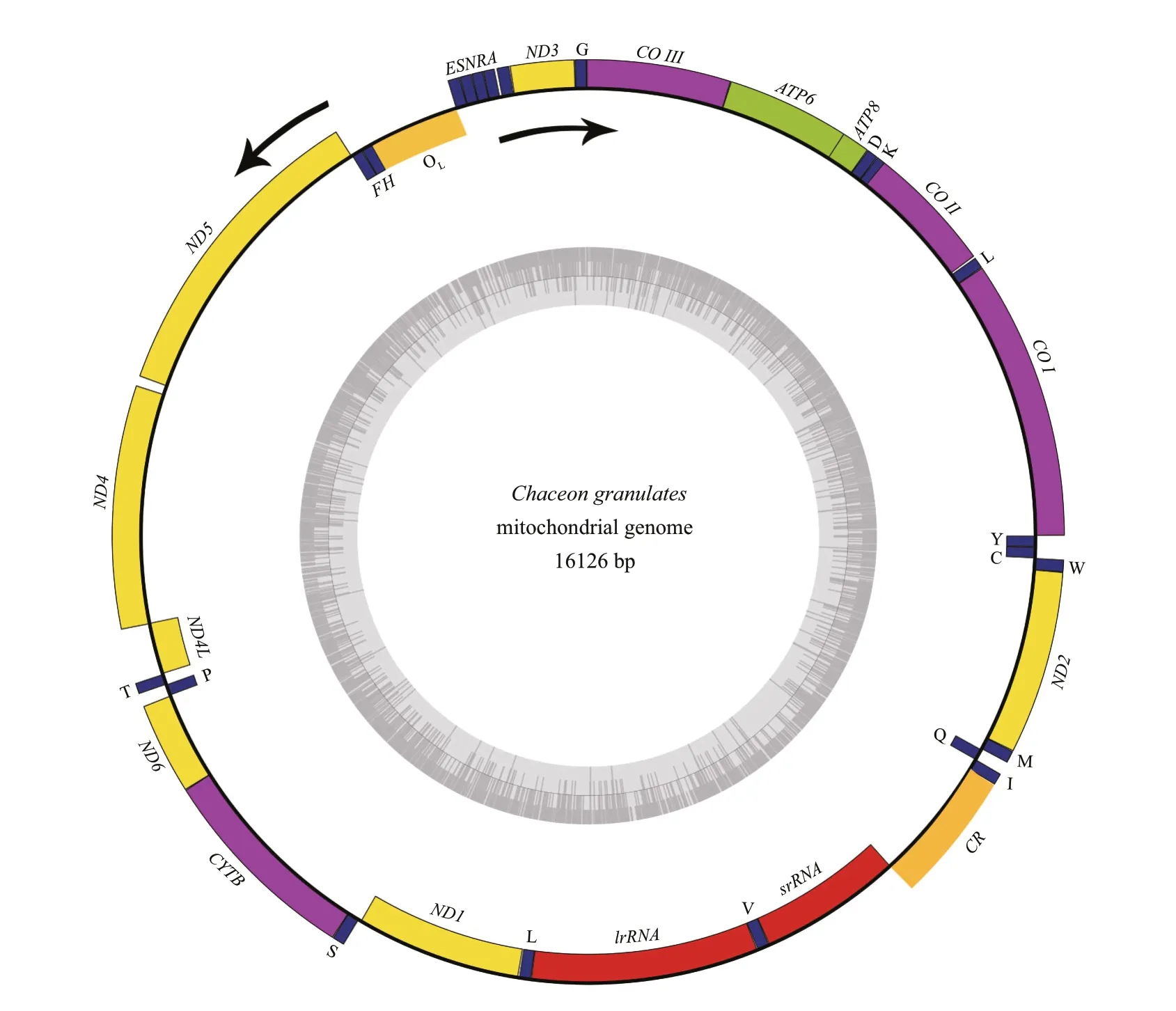
Fig.1 Graphical map of the complete mitogenome of C. granulates
The selective pressure on the crab mitogenome was evaluated using CODEML in the PAML package.Two diff erent tree-building methods were used because the CODEML likelihood analysis is sensitive to the tree-topology. The two-ratio and free-ratio model (M1 model) was used for the mitogenome analysis. The branch-site model was used to determine whether these genes have undergone positive selection on foreground branches. Bayes Empirical Bayes(BEB) analysis was used to calculate the Bayesian posterior probability of the positively selected sites.
2.5 Phylogenetic analysis
To illustrate the phylogenetic relationships among crabs, the complete mitogenomes of 23 Decapoda species were downloaded from the GenBank database(Supplementary Table S2).Harpiosquillaharpax(Stomatopoda) was selected as an outgroup. The concatenated nucleotide sequences of 13 energy pathway PCGs were aligned using Clustal X with the default settings. The maximum likelihood (ML)method was used to analyze the phylogenetic trees.The GTR+I+G model was selected as the best nucleotide substitution model by using ModelTest 3.7(Posada and Crandall, 1998). The ML analysis was performed with MEGA 5.1 with 1 000 bootstrap replicates.
3 RESULT AND DISCUSSION
3.1 Genome organization
The mitochondrial DNA of the cra b is a circular molecule, with 16 126 bp in length. The sequence alignment showed the most identical (98%) with anotherC.granulates(NC_AB769383) uploaded before. Like most other metazoan mitogenomes(Green and Reed, 1998; Boore, 1999; Bernt et al.,2013), the present crab mitogenome contains 13 PCGs, 22tRNAgenes, 2rRNAgenes, and a control region (CR) (Table 1, Fig.1). Of the 37 genes, 23 genes were encoded by the heavy strand (H-strand).The gene order and orientation were identical to other decapods (Yamauchi et al., 2003; Miller et al., 2005;Liu and Cui, 2010; Ma et al., 2013). Eight geneoverlaps and eleven intergenic spacers were observed.Except for a 527-bp untranslated region betweentRNA GluandtRNA His, the gene overlaps and intergenic spacers were similar to those in other sequenced Decapod crabs. The complete mitochondrial DNA sequence was deposited in the GenBank database under the accession number KU507298.

Table 1 Gene structure of C. granulates mitogenome
The metazoan mitogenome usually has a strand-specif ic bias in nucleotide composition (Hassanin et al., 2005). In the present study, the nucleotide composition had a bias toward A and T. The overall A+T content in the H-strand was 69.19%(Supplementary Table S3), which is within the range of the rates observed in other Decapods (Shen et al.,2007). InC. granulatesmitogenome, similarly to other crabs (Liu and Cui, 2010; Ma et al., 2013), the highest A+T content was detected in the CR (77.58%),and the lowest A+T content was found in the 13 PCGs(67.03%). The AT-skew and GC-skew showed a similar tendency in decapods (Liu and Cui, 2010; Ma et al., 2013). The AT-skew (-0.039) and the GC-skew(-0.205) in the H-strand were negative, thus indicating a preference for A and C inC. granulatesmitogenome.
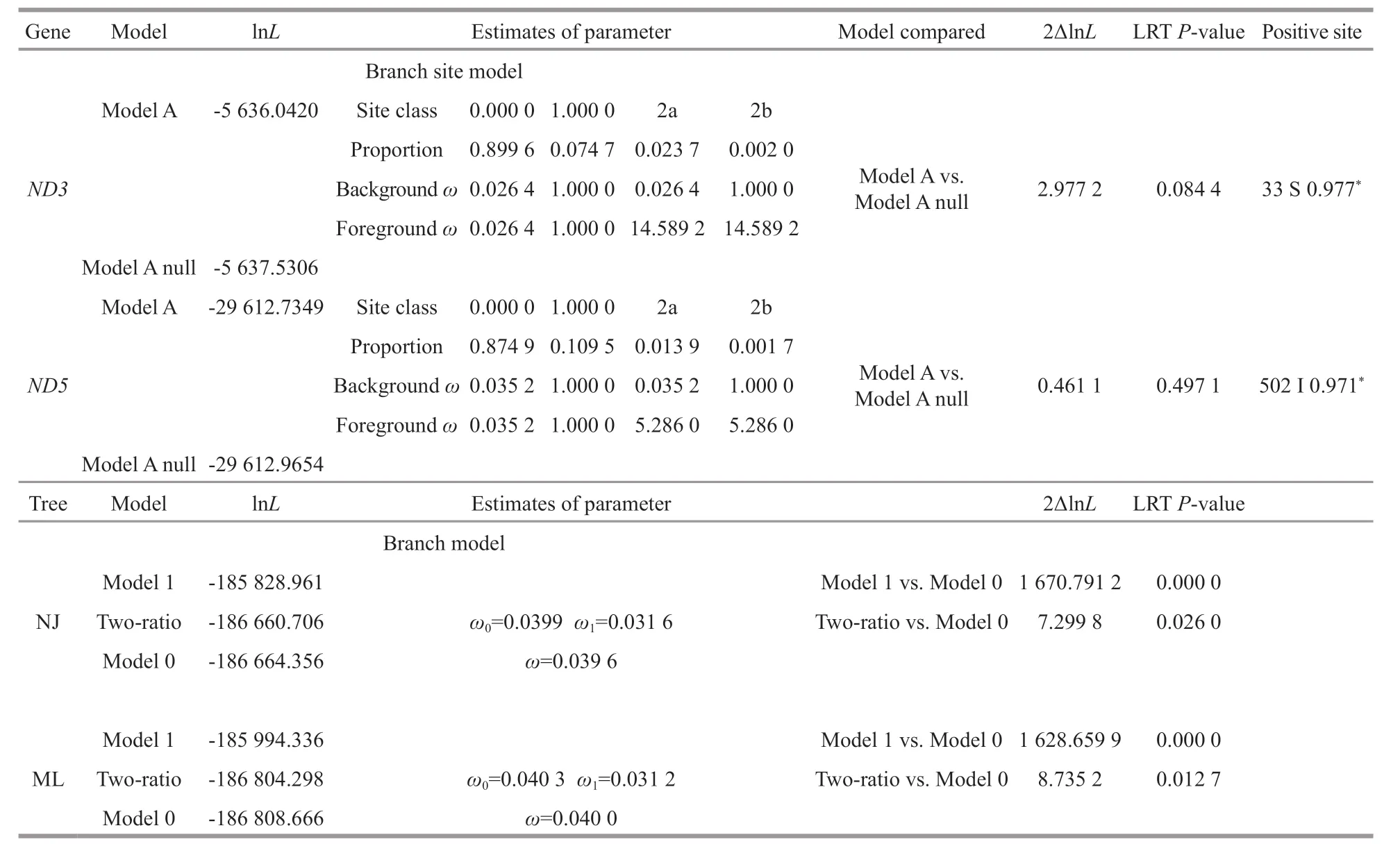
Table 2 Selective pressure analyses of the mitochondrial genes of crabs
3.2 Protein-coding genes
In total, 13 PCGs were identif ied in theC.granulatesmitogenome (Table 1). These genes spanned 11 204 bp and encoded 3 724 amino acids.The arrangements of PCGs in several Brachyura species were consistent (Fig.2). InC. granulatesmitogenome 9 PCGs were encoded by the H-strand(Fig.1). Except forND4, which was initiated by a rare initiation codon (GTG), the remaining genes started with the typical ATN codons (ATG or ATT). ATG was the most common initiation codon, which initiated 9 of the 13 PCGs (Table 1). Three kinds of termination codons (TAA, TAG, and an incomplete stop codon T)were observed. The most common termination codon was TAA. In the metazoan mitogenome, incomplete termination codons produce functional termination codons through polycistronic transcriptional cleavage(Ojala et al., 1981; Boore, 2001). In the present study,4 PCGs were terminated by the truncated termination codon T, in which the missing nucleotides may be produced through post-transcriptional polyadenylation(Ojala et al., 1981).
The codon usage in the 13 PCGs revealed that UUA (Leu), AUU (Ile), and UUU (Phe) were the three most frequently utilized codons inC. granulatesmitogenome (Supplementary Table S4, Fig.3a). The codon distribution patterns in 11 Brachyura and 1 Anomura species were highly conserved (Fig.3b).
3.3 Positive selection analysis
The selective pressures imposed on the crab mitogenomes were evaluated using CodeML in the PAML package (Table 2). The result showed that the mitochondrial sequences in the genusChaceonexperienced diff erent selective pressures compared with shallow species, and signif icant evidence of positive selection was detected in two sites (33S inND3and 502I inND5) ofC. granulates(BEB value >0.95).
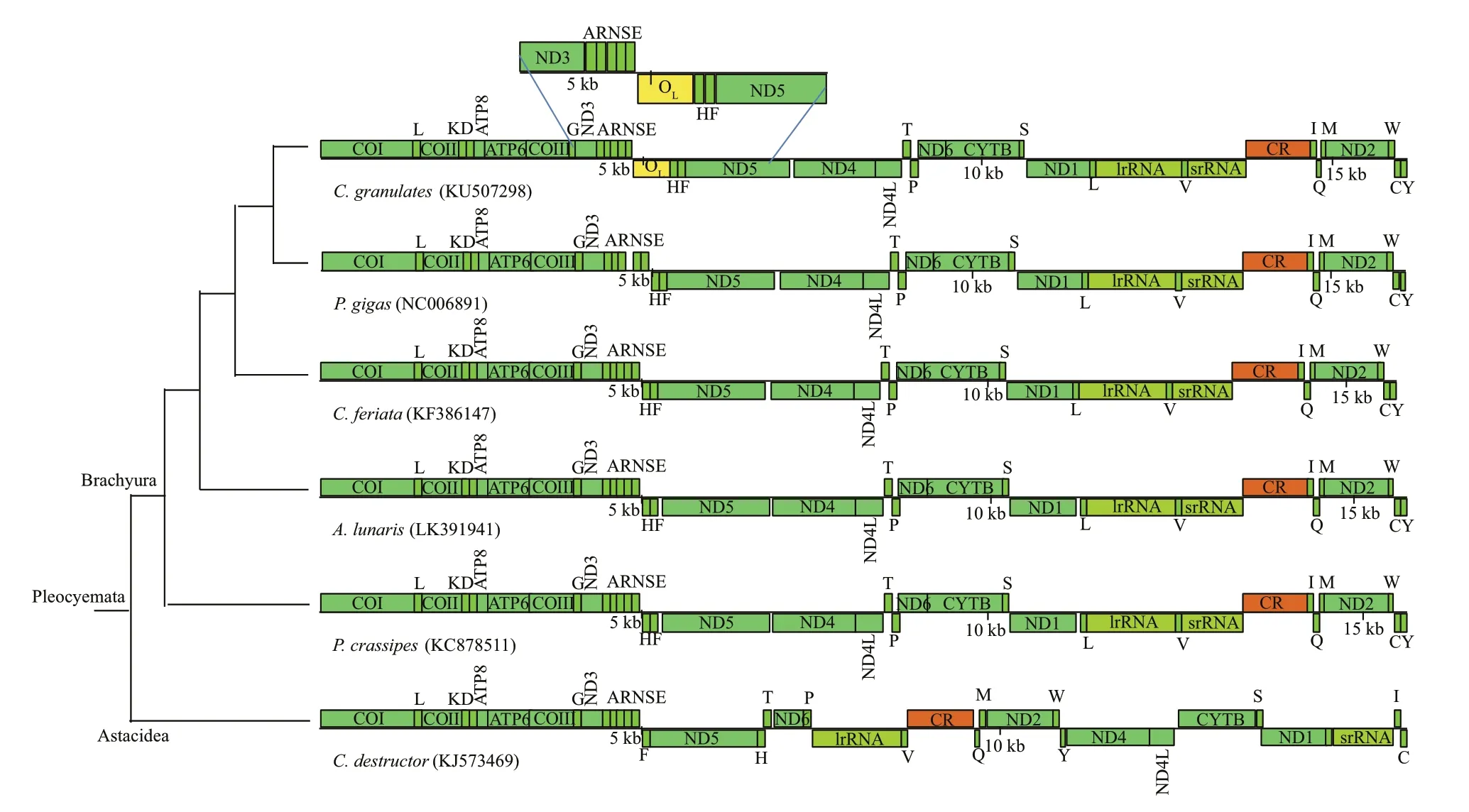
Fig.2 Linearized schemes of the mitochondrial gene arrangements in Brachyura C. destructor (Astacidea as an outgroup)
During the evolution, protein positive selection may act in very short episodes, and the eff ect may be only on a few sites along lineages in the phylogeny(Sun et al., 2018). The severe environmental conditions can aff ect the metabolism and direct selection of mitochondrial DNA (Ning et al., 2010). As the key components of energy metabolism and various biosynthetic pathways (Green and Reed, 1998;Newmeyer and Ferguson-Miller, 2003), the mutations in mitochondrial protein-coding genes may inf luence the electron transport chain and further inf luence the energy metabolism and other biosynthetic pathways(Sun et al., 2018). Many studies have recently shown evidence for positive selection acting on the mitochondrial genome, emphasizing its potential role in adaptive divergence and speciation (Jacobsen et al.,2016). Organisms living under extreme environmental conditions, there must be certain modif ications in energy metabolism to adapt to environment (Zhang et al., 2017a, b; Sun et al., 2018). As a proton pump, the NADH dehydrogenase complex is the f irst and the largest enzyme complex in the respiratory chain. Thus,it plays an important role on the adaptive evolution in many species. In Alvinocarididae lineages, the residues with the highest number of positively selected sites are within nad1-5 (Sun et al., 2018), and is considered associated with deep sea hydrothermal vents adaptation.ND2andND6were found to be under positive selection in mitogenome analysis of Chinese snub-nosed monkeys, it was known to be related to adaptive changes high altitude and cold weather stress(Yu et al., 2011). In Tibetan horses,ND6was found to be under positive selection, which was associated with high altitude living adaptation (Ning et al., 2010). In whitef ish (Coregonusspp.),ND2showed a highly elevated dN/dS ratio, which was considered to drive the adaptive evolution (Jacobsen et al., 2016). The mitochondrial whole-genome comparison study in 40 Tibetan and 50 Han Chinese people provide clues for the existence of adaptive selection for theND2in Tibetans, which likely contributed to adaptation to their specif ic geographic environment, such as high altitude (Gu et al., 2012).
In this study, two sites of NADH dehydrogenase complex presented signif icant evidence of positive selection. The result supports the hypothesis of adaptive evolution in the mitogenome of deep-sea environment. The mutations in these subunits may inf luence the effi ciency of the proton-pumping process. They may potentially have functional implications to the energy metabolism. Considering the living environment, the mutations in NADH dehydrogenase complex may contribute to the adaptation to deep-sea environment.
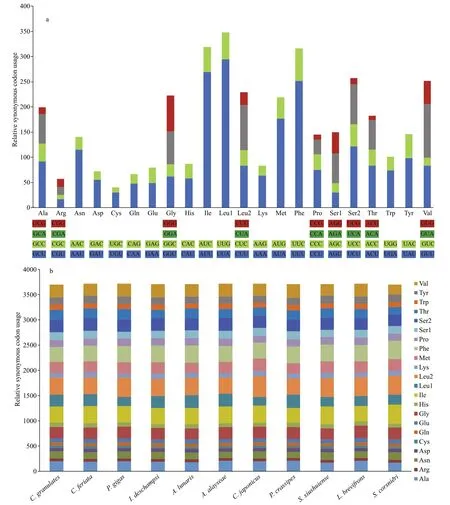
Fig.3 Codon usage in crabs
3.4 tRNA genes and rRNA genes
In total, 22 tRNAs were identif ied inC. granulates(Table 1), with sizes ranging from 62 to 72 bp. Of the 22tRNAgenes, 14tRNAgenes were located on the H-strand. Except fortRNA Ser(AGA), all 21 tRNAs folded into a cloverleaf secondary structure (Supplementary Fig.S1). ThetRNA Ser(AGA)presented an unusual secondary structure lacking the stem-loop structure in the DHU arm, which is observed in other crabs (Liu and Cui, 2010). In addition, 4 unmatched base pairs were observed in theC. granulatesmitogenome. The tRNACyscontained an A-A mismatch on the DHU arm, but the remaining 3 mismatched tRNAs occurred in the amino acid acceptor arm. Such stem mismatches appear to be a common phenomenon in mitochondrial tRNAs in many species (Miller et al., 2005; Liao et al., 2010; Jiang et al., 2013; Wang et al., 2016) and are probably corrected through a post RNA-editing mechanism (Lavrov et al., 2000).
Like most metazoan mitogenomes, 2rRNAgenes(lrRNA andsrRNA) were present inC. granulates(Table 1). InC. granulatesmitogenome, thelrRNA andsrRNAgenes were located on the L-strand and contained 1 326 bp and 834 bp, respectively. The A+T contents were 74.53% and 73.50%. ThelsRNAgene was located between thetRNALeu(CUA)andtRNAValgenes. ThesrRNAgene was located between thetRNAValgene and the putative CR.
3.5 Non-coding regions
Thirteen non-coding regions were identif ied in the mitogenome ofC. granulates(Table 1). The longest intergenic region was the putative CR. It is considered essential for mitochondrial genes transcription and replication in vertebrates (Fernández-Silva et al.,2003). It is usually considered the most variable portion of the mitogenome (Marshall and Baker,1997). The nucleotide composition of the CR(H-strand) was 314 A (43.73%), 243 T (33.84%), 103 C (14.35%), and 58 G (8.08%). It showed a similar tendency in Brachyuran crabs. However, the nucleotide composition and length variations were evident among crabs. The nucleotide alignment of the CR in Brachyuran crabs showed low homology,which was also conf irmed inCharybdisjaponica(Liu and Cui, 2010). The length of the CRs in crabs were range from 514 bp to 1 435 bp (Supplementary Table S3). The result is consistent with the studies in other crustaceans (Valverde et al., 1994; Umetsu et al.,2002).
In addition to the largest CR (718 bp) in the mitogenome ofC. granulates, another non-coding sequence (527 bp) was detected, which is a unique insertion that just detected inChaceoncrabs.Moreover, the sequence analysis of the 527 bp noncoding sequence exhibited several typical “CR-like”characteristics. Firstly, similar to the def ined CR, the non-coding sequence was much larger than the remaining 11 non-coding regions, which ranged from 1 bp to 50 bp. The result is identical to the CR observed inPocillopora(Flot and Tillier, 2007).Secondly, both of the 718-bp CR (H-strand) and the 527-bp genetic fragment (L-strand) showed similar nucleotide composition. The nucleotide composition of the 527-bp non-coding region was 229 A (43.45%),172 T (32.64%), 45 C (8.54%), and 81 G (15.37%).Both parts showed almost the same A+T content rate,which is higher than that in the other regions in the mitochondrial DNA. High rate of A+T is a common feature in all organisms except primates (Sbisa et al.,1997). Thirdly, although the CR in Brachyuran crabs showed low homology; several conserved motifs were identif ied in both parts and other several CRs of Brachyuran crab (Supplementary Fig.S2c). The“TACAT” motif, which is observed in some f ish (Guo et al., 2003), was found inC. granulates(Supplementary Fig.S2a & b). The “G(A)nT” motif,which was present in the 3′ f lanking sequences of mammalian, amphibian, and f ish mitochondrial L-strand replication origins, and showed an universal conservation and functional importance related to replication origins (Zhang et al., 1995), was also detected in the 527-bp non-coding region(Supplementary Fig.S2a & b). Thus, we presumed that the 527-bp non-coding region could be a regulatory element in the present study.
Further structural analyses showed that the secondary structure of the 527-bp non-coding region presented several stem-loop structures (Supplementary Fig.S3), which is a characteristic feature of the origin of light-strand replication (OL) in vertebrates (Clayton,1991). Moreover, the locus of the 527-bp non-coding region in mitogenome was identical to the OLin most vertebrates. In most vertebrates, the OLwas located in a cluster of tRNA genes (between tRNAAsnand tRNACys), which known as the WANCY region(Kawaguchi et al., 2001; Jin et al., 2015). Therefore,we concluded the 527-bp non-coding region could be the OLofC. granulates. At present, among Brachyuran crabs, such non-coding region (OL) was detected in mitogenomes of deep-seaChaceoncrabs only (Fig.2).
In the study of deep-sea hydrothermal vents and cold seepsAlvinocarislongirostris, seven putatively duplicated gene clusters ofcytochromeP450swere expressed diff erentially, which is considered to contribute to the adaptation to harsh conditions (Hui et al., 2018). Considering the OLcan inf luence the replication and transcription of mitochondrial genes,we speculated the OLcould participate in the regulation of mitochondrial genes expression. Thus,it could be indirectly involved in adjusting mitochondrial energy metabolism to adapt to the deep-sea environment.
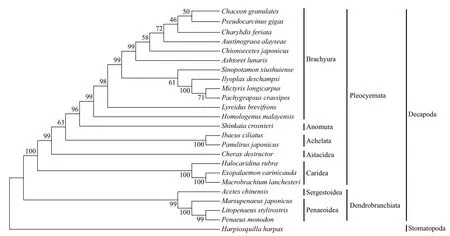
Fig.4 Phylogenetic analysis
3.6 Phylogenetic analyses
We performed a phylogenetic analysis of the crabs based on the nucleotide datasets of 13 mitochondrial energy pathway PCGs (Fig.4). The complete mitochondrial genome ofC. granulatesprovides well resolved molecular phylogeny of the Decapoda. The phylogenetic tree supported the hypothesis that Decapoda was reorganized into the Pleocyemata and Dendrobranchiata suborders (Burkenroad, 1981), a result consistent with the decapod phylogeny based on other molecular data (Tsang et al., 2008). Together with 11 other species,C. granulatesf irst formed a monophyletic group with a 100% bootstrapping value,which clustered in the Brachyura clade. Anomura,Achelata, Astacidea and Caridea composed the other clades in the Pleocyemata group. The Brachyura clade showed a close relationship with the Anomura clade, as has been shown in another study (Tsang et al., 2008;Liu and Cui, 2010; Ma et al., 2015).
4 CONCLUSION
The complete mitogenome of a deep-sea crab,C.granulates, was characterized and compared with other shallow decapods. The complete mitogenome ofC. granulatesis a typical circular molecule, with 16 126 bp in length. The genetic composition, order,and orientation are similar to those of closely related crabs, belonging to the Brachyura branch clade of Decapoda. Several genes showed strong evidence of positive selection, in genusChaceonof the mitogenomic analysis, andND3andND5were determined to be under positive selection with a BEB value >95%, by using the branch-site model in CODEML.C. granulatesmitogenome possesses a unique “OL” that is not present in shallow crabs.Therefore, given that the OLin mitogenomes is essential for mitochondrial genes expressional regulation, as well as positive selection genes, we speculated that two special gene features might play important roles in regulating the mitochondrial energy metabolism that could involve in the adaptation to the deep-sea conditions. The data presented in this study may shed a light on the knowledge of mitogenomic adaptation in the deep-sea environment.
5 DATA AVAILABILITY STATEMENT
The authors declare that all data supporting the f indings of this study are available within the article and its supplementary f iles.
 Journal of Oceanology and Limnology2020年2期
Journal of Oceanology and Limnology2020年2期
- Journal of Oceanology and Limnology的其它文章
- Contribution of surface wave-induced vertical mixing to heat content in global upper ocean*
- Upper ocean response to typhoon Kujira (2015) in the South China Sea by multiple means of observation*
- Inf luence of simulating deep-sea environmental factors on cathodic performance of seawater battery*
- Adsorption characteristics of chitooligosaccharides onto activated charcoal in aqueous solutions*
- Eff ects of hypoxia on survival, behavior, and metabolism of Zhikong scallop Chlamys farreri Jones et Preston 1904*
- Distinct inf luence of trimethylamine N-oxide and high hydrostatic pressure on community structure and culturable deep-sea bacteria*