
Genetic structure and demographic histories of two sympatric Culter species in eastern China*
2020-03-19 12:31XIONGYingLIWeiYUANJingZHANGTanglinLIZhongjieXIAOWuhanLIUJiashou
XIONG Ying , LI Wei YUAN Jing ZHANG Tanglin LI Zhongjie XIAO Wuhan ,LIU Jiashou
1 State Key Laboratory of Freshwater Ecology and Biotechnology, Institute of Hydrobiology, Chinese Academy of Sciences,Wuhan 430072, China
2 University of Chinese Academy of Sciences, Beijing 100049, China
Abstract Geographic isolation is a key factor in shaping the genetic structure of many f ish species.Two sympatric species, Culter alburnus and C. mongolicus, are economically important f ish that are widely distributed in China and have been recently used as new aquaculture species. We used the mitochondrial DNA control region (CR) as a marker to investigate the genetic structure of the two species. Phylogenetic analysis revealed two major lineages (Lineages I and II) that were highly consistent with geographical patterns for C. alburnus and C. mongolicus. Based on genetic distance, the Zhujiang (Pearl) River Basin (ZRB)populations potentially represented a cryptic subspecies, which might be diff erentiated as the result of strict geographic isolation, an earlier diverged event, or peripheral areas. Genetic diversity analysis suggested that the Changjiang (Yangtze) River Basin (CRB) populations are located at, or near the core region of their origination, not only due to the larger population size at the CRB, but also their habitat diversity and suitability for its survival, and the genetic diversity diff erences among basin populations were signif icant for the two species. Moreover, demographic analysis indicated that the two Culter species and most populations had undergone a period of population expansion during warm interglacial periods. However, the C. alburnus population of Huaihe River Basin (Weishan Lake) exhibited diff erent patterns during the interglacial period,which may due to the latest diverged time of HRB and Weishan Lake located at the permafrost. Notably,the ZRB (in Songtao Reservoir) C. mongolicus population showed no genetic diversity and had a unique haplotype, which could be treated as a special gene pool for species conservation. In summary, geographic isolation is most likely responsible for the two Culter species distribution patterns.
Keyword: Culter species; drainage basin; genetic diff erentiation; geographic isolation
1 INTRODUCTION
The distribution of f ish is strictly constrained by the drainage basin because of geographic isolation. In association with the development of river systems and climatic f luctuations, freshwater f ish may undergo vicariance, resulting in genetic diversif ication and even allopatric speciation (Mayr, 1942; Rundle and Nosil, 2005; Tedesco et al., 2012; Yu et al., 2014).Freshwater f ish are more susceptive to the physicochemical environment than marine f ish, so their distribution patterns are thought to be more related to climate change (Comte et al., 2013).Phylogeography has provided new insights to evaluate the inf luence of historical events on the genetic structure and composition of modern populations,which has revolutionized population genetics and historical ecology (Avise et al., 1987). Moreover, the phylogenetic study of widespread freshwater f ish is an excellent indicator for geographic events.
Gene f low among freshwater f ish populations within the same catchment is common, but dispersal among diff erent catchments and drainage basins is typically restricted because of the lack of hydrological connectivity (Mesquita et al., 2005; Huey et al.,2008). However, freshwater f ish often inhabit multiple isolated drainage basins, implying dispersal among drainages has been possible in the past (Huey et al.,2011). In China, the colonization of drainages by freshwater f ish can be explained by a variety of mechanisms. These include catchment rearrangements through erosion and uplift, climatic-induced f luctuations in sea level (Li, 1981), and anthropogenic translocation (Xiong et al., 2015).
The glacial periods of the Quaternary were a major factor in shaping contemporary distributions of extant f lora and fauna (Provan and Bennett, 2008). Fossil and pollen record evidence also suggests that most of the biota, particularly temperate species, survive glacial maxima in lower latitude refugia (Bennett et al., 1991). Many studies have indicated that Quaternary glacial climatic oscillations are too close to explain lots of deep genetic divergences observed nowadays (Huang, 1982; Miralles and Carranza,2010; Yu et al., 2014). Glacial impacts were greatest in Eurasia, North America, and Antarctica, where glacial ice covered major portions of their surfaces(Hewitt, 2003). China was also aff ected by the glacial cycle during the Quaternary Period. In the cold periods, mainland China was mainly inf luenced by alpine glaciers and cold air. During the warm periods,the mainland climate was dominated by warm humid air masses, which led to the retreat of alpine glaciers(Yu et al., 2014).
Cyprinids are the largest extant family of vertebrates and the most diverse freshwater f ish family in Asia. The genusCulterconsists of nine species and subspecies that are conf ined to Asia,particularly China, Vietnam, Korea, Mongolia, and Russia (Chen, 1998). Several studies have investigated phylogenetic and biogeography of theseCulterspecies (Wang et al., 2008; Qi et al., 2013,2015; Yang et al., 2016), however, the sample sizes were small and geographic distances were limited.Therefore, a much larger and more intensive survey is needed to determine the genetic structure ofCulterpopulations. The topmouth culter (Culteralburnus)and Mongolian redf in (C.mongolicus) are the largest carnivorous f ish of theCultergenus (Chen, 1998).They are commercially important freshwater f ish that widely coexist in larger rivers, lakes, and reservoirs of China. Their recent use for commercial aquaculture in some provinces of China has seen rapid development (Fan et al., 2008), but wild populations of both species have declined so quickly that conservation of their genetic diversity has become a very important issue.
Our current understanding suggests that extantCulterspecies originated from the Pliocene epoch in the River Plain of Eastern China (RPEC) according to fossil records (Liu and Su, 1962; Wang, 2013). The Bohai basin is a third-order reservoir constrained by the Miaodao Uplift that was formed by subsidence during the Cenozoic Era (Allen et al., 1997; Hu et al.,2001; He and Wang, 2003). It experienced a threestage evolution from the early Pliocene (3.7 million years ago, Ma) to a marine system in the late Pleistocene (0.13 Ma) (Yi et al., 2016). Fish spread from the RPEC along the Liaohe River and Nenjiang River to Heilong River Basin (LRB) during the Pleistocene. Analysis of the natural distribution of extant f ish species in the Zhujiang (Pearl) River Basin(ZRB) suggested that the southward diff usion of East Asian taxa might be caused by the uplift of the Yunnan plateau in the late Pliocene. Hainan Island originally belonged to the ZRB, and was still connected to the South China mainland until the early Quaternary Holocene (10 000 years ago) (Wang et al., 2012).Today,Culterspecies are widely distributed in Eastern China, especially in the Changjiang River Basin(CRB), Huaihe River Basin (HRB), LRB, and ZRB.We hypothesize thatCulterspecies were subject to drainage basin isolation, so applied the isolation by distance (IBD) model in our present study. Under the population genetics background, this is the process of generating genetic structures through geographically restricted gene f low because random genetic drift is occurring locally (Hardy and Vekemans, 1999).
Here, we employed the mitochondrial DNA control region (CR) to test hypotheses on the drivers of biogeographic process in two sympatric species,C.alburnusandC.mongolicus. Large-scale sampling allowed us to test hypotheses on the evolutionary history and population demographics of these species to determine if Pliocene and Quaternary climatic f luctuations resulted in their genetic diversity in China. We also evaluated the genetic diversity,population genetic structure, and population demographic history ofC.alburnusandC.mongolicusand determined whether IBD played a role in shaping the structure ofC.alburnusandC.mongolicus.Finally, we assessed the level of genetic diversity across all current populations and proposed suitable conservation plans.
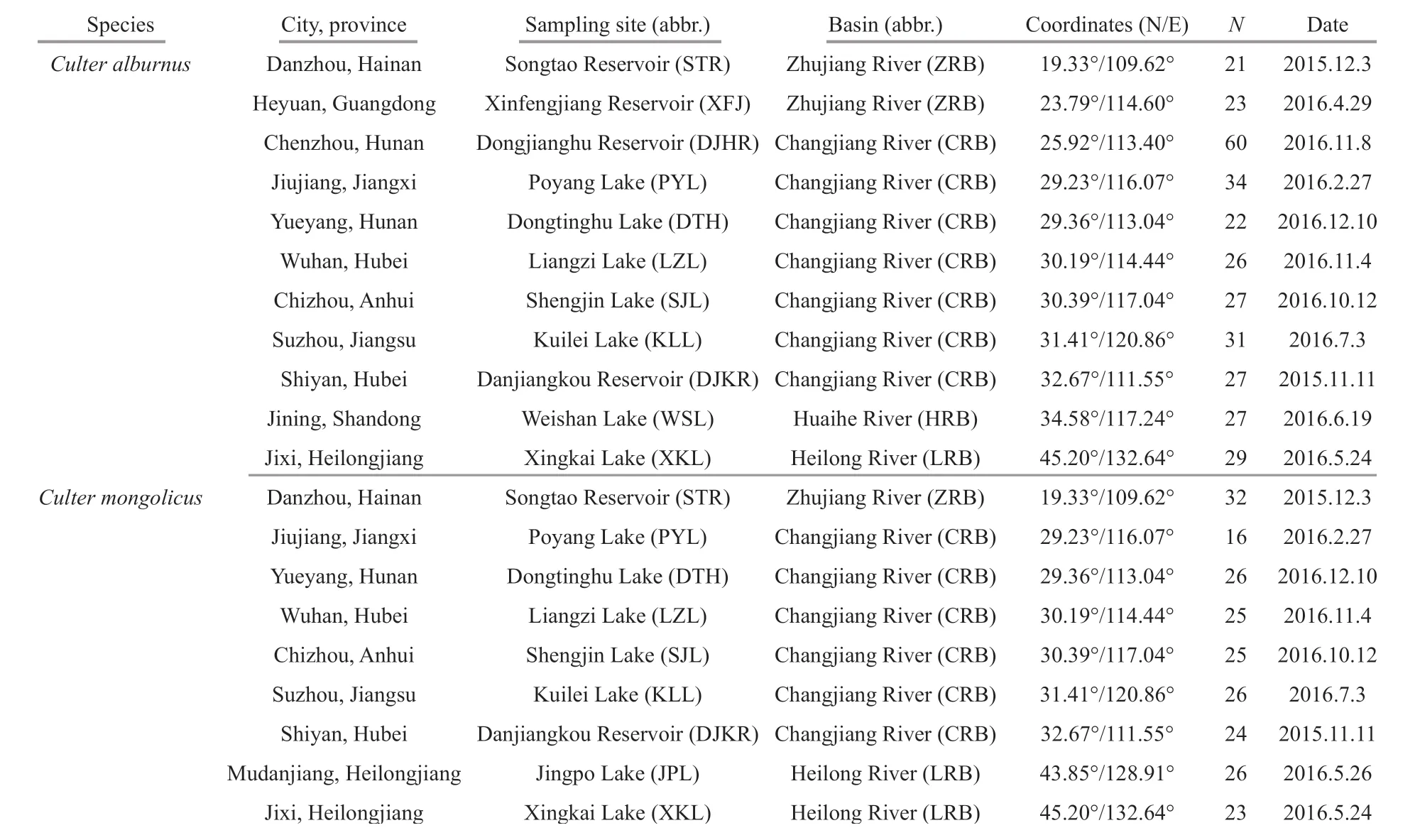
Table 1 Sample site information
2 MATERIAL AND METHOD
2.1 Samples collection
A total of 333 wildC.alburnusspecimens and 225 wildC.mongolicusspecimens were collected using three layer gill net from 12 lakes and reservoirs in China between November 2015 and March 2017.Sample information is provided in Table 1 and sampling sites are mapped in Fig.1. Caudal f in tissue samples used for genomic DNA extraction were preserved at 4°C and in 100% ethanol and deposited in the Institute of Hydrobiology, Chinese Academy of Sciences.
The Holocene is the latest phase of the geological era when the biological composition was almost identical to that of the modern era (Yan, 2006). In this study, the Songtao Reservoir (STR) on Hainan Island was classed as the ZRB because Hainan Island once belonged to the ZRB, and separated from the South China mainland during the late Quaternary (Zeng and Zeng, 1989).
2.2 DNA extraction, PCR amplif ication and sequencing
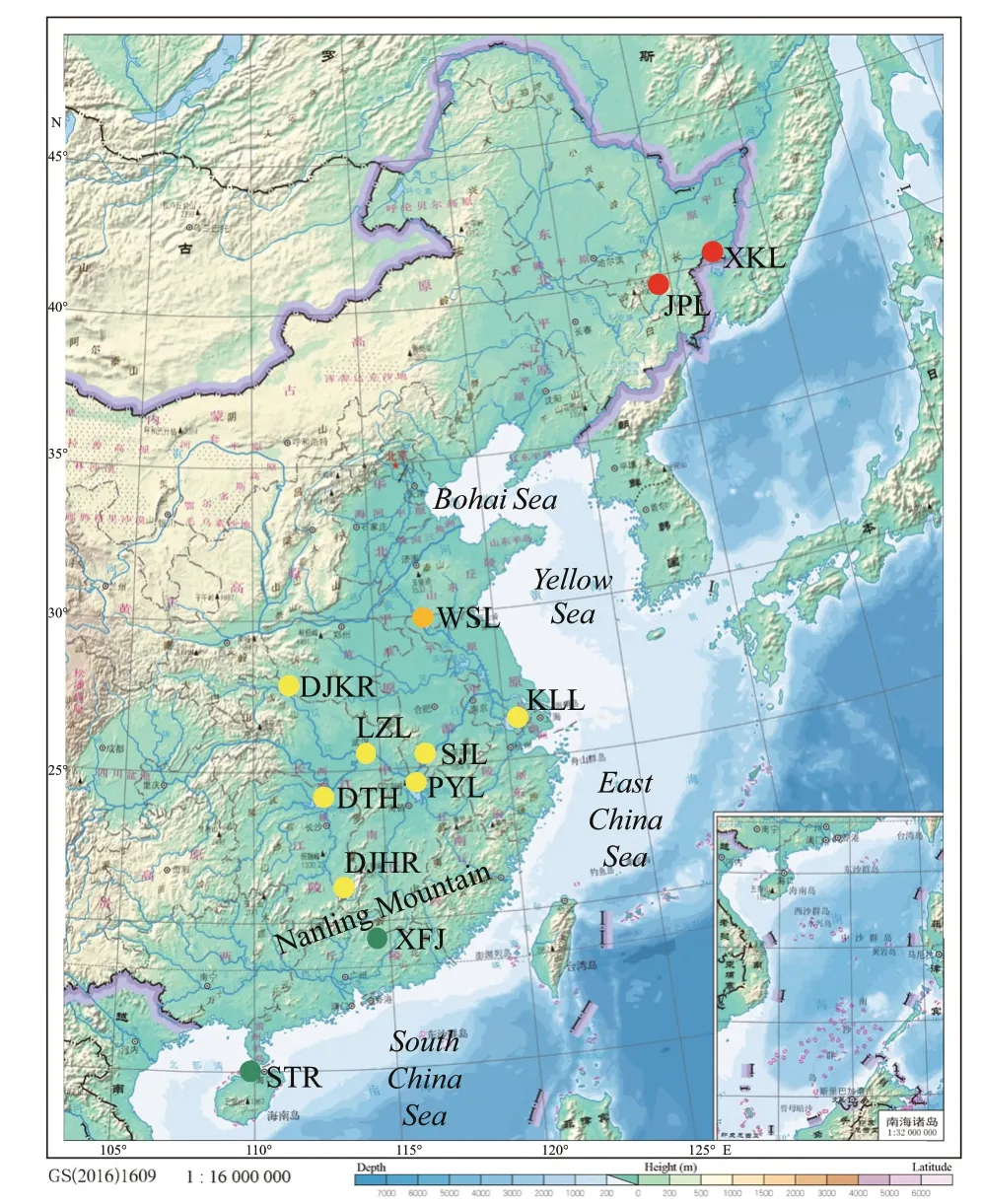
Fig.1 Map of sampling localities for Culter alburnus and Culter mongolicus populations
Total genomic DNA was extracted from topmouth culter and Mongolian redf in tissue using phenolchloroform extraction (Russell and Sambrook, 2001).The mitochondrial CR was amplif ied using the universal primers MitD1-F: 5′-CACCCYTRRCTCCCAAAGCYA-3′, and MitD1-R: 5′-GGTGCGGRKACTTGCATGTRTAA-3′ (Huang et al., 2007; Cheng et al., 2011). Each f inal reaction volume of 40 μL contained 26.4 μL of sterilized ultrapure water, 4 μL 10× Ex-taq buff er, 4 μL of Mg2+(25 mmol/L), 1.6 μL of dNTP-Mix (2.5 mmol/L), 0.4 μL Extaq (5 U/μL),0.8 μL of each primer (10 mmol/L), and 2 μL of DNA template (approximately 100 ng/μL). PCR amplif ications were performed using a T100 Thermal Cycler with a 5 min at 94°C, followed by 5 cycles at 94°C for 30 s,58°C for 30 s, and at 72°C for 1 min, then 35 cycles at 94°C for 30 s, 60°C for 30 s, 72°C for 1 min, with a f inal elongation step at 72°C for 7 min. Amplif ied PCR products were purif ied by 1.0% low-melting agarose gel electrophoresis and sequenced in both directions using primers F-SEQ-0510: 5 ′-TGCATTTGGCTTCTATCTCAGG-3′ and R-SEQ-0510:5′-CCTACTTAAACTCAAGCAAGG-3′ and the ABI PRISM® 3730 DNA Analyzer (Applied Biosystems,Life Technologies, Shanghai, China) by the Tsing Ke Commercial Sequencing Service (Wuhan, China).
2.3 Phylogenetic analysis
The generated sequences were aligned against previously published sequences for the same genetic region, and trimmed to the same length using the software package MEGA 7.0 (Kumar et al., 2016).
Phylogenetic relationships based on CR haplotypes were estimated using Bayesian Inference (BI) and Maximum Likelihood (ML) methods. BI analysis was performed using MrBayes 3.2.6 (Ronquist et al.,2012). The best-f it nucleotide substitution models forC.alburnusandC.mongolicuswere TrN+G and HKY+I+G, respectively, which was based on the Bayesian Information Criterion in jModelTest 2.1.7(Darriba et al., 2012). Four simultaneous Markov chain Monte Carlo (MCMC) methods were run for 20 million generations sampling once every 1 000 generations, two independent parallel analyses methods were used. The average standard deviation of split frequencies was less than 0.01 and the potential scale reduction factor value was close to 1. The f irst 20 000 sampled trees were discarded as burn-in and 50% majority-rule consensus trees was calculated from remaining trees with posterior probability values for each node. Support for clades is expressed as posterior probabilities.
The MLs for phylogenetic analysis were reconstructed in PhyML 3.0 (Guindon and Gascuel,2003) with 1 000 bootstrap replicates. The most suitable model of DNA substitution, TrN+I+G forC.alburnusand HKY+I+G forC.mongolicus, was determined by ModelTest 3.7 (Posada and Crandall,1998). Grass carp (Ctenopharyngodonidellus)(GenBank: KJ614563.1) andHemiculterleucisculus(GenBank: AF494361.1) were set as outgroups.
We constructed haplotype networks using Medianjoining network analysis by Network 4.6 (Bandelt et al., 1999) to visualize relationships among all haplotypes.
2.4 Divergence time estimates
We employed the relaxed molecular clock method incorporated in BEAST 1.8.4 to estimate the divergence time of each lineage (Drummond and Rambaut, 2007). Calibration information was according toC.mongolicus-like fossil records detected in the Pliocene (Liu and Su, 1962; Chen,1998). The means values (4±1.9 Ma) of the normal distributions were used in the analysis. The Yule process was used as the tree topology prior, and the TrN and HKY substitution models were employed forC.alburnusandC.mongolicus, respectively. Posterior distributions of node ages were performed by MCMC for 20 million generations with sampling every 1 000 trees, and the f irst 10% was treated as burn-in. The eff ective sample size was used to determine the Bayesian statistical signif icance of each parameter in Tracer 1.6 (Rambaut and Drummond, 2013).Summarized and annotated phylogenetic trees with a molecular clock constraint yielded by BEAST were displayed by FigTree 1.4.3 (Rambaut, 2009).
2.5 Genetic diversity and population structure
Genetic diversity is ref lected in the measures of the number of haplotypes (n), haplotype diversity (π) and nucleotide diversity (h) (Nei, 1987), using DnaSP 5.10 (Librado and Rozas, 2009) software calculate values of each population. The mean genetic distance within and between populations were calculated according to the Kimura 2-parameter (K2P) model(Kimura, 1980) as implemented in MEGA7, standard errors were estimated by bootstrapping using 10 000 replicates. Pairwise genetic diff erentiation between populations was estimated usingF-statistics (FST)(Weir and Cockerham, 1984) with 10 000 permutations, based on the distance method in Arlequin 3.5 (Excoffi er and Lischer, 2010). To investigate signif icant genetic structure between populations, we performed a two-level hierarchical analysis of molecular variance (AMOVA) using Arlequin. Signif icance was assessed by 10 000 random permutations. Gene f low (Nm) was evaluated from GammaST (Nei, 1982), andFST(Hudson et al.,1992) was evaluated using DnaSP 5.10.
Mantel tests were used to test the relationships between pairwise values ofFSTand the geographical distance. Geographic distances among populations were estimated using Google Earth 7.3.0.
2.6 Demographic history
The population demography for topmouth culter and Mongolian redf in populations were examined using three approaches. First, Tajima’sD(Tajima,1989) and Fu’sFs(Fu, 1997) were applied to check the evidence of neutral evolution in Arlequin 3.5(Excoffi er and Lischer, 2010). In the neutrality test,we assessed signif icance by generating null distributions from 10 000 coalescent simulations.
Second, the demographic history was investigated by comparing mismatch distributions in each basin population sample using DnaSP 5.10 and Arlequin3.5.The time of possible population expansions (t) was estimated via equationτ=2ut(Nei and Tajima, 1981;Rogers and Harpending, 1992), whereuis the mutation rate for each sequence and each generation.The value ofuwas calculated byu=2μk, whereμis the mutation rate per nucleotide andkis the number of nucleotides in the analyzed fragment. The approximate time of expansion was calculated by multiplyingtby the generation time (C.alburnus:3 years for CRB, HRB, and ZRB, 4 years for LRB,and 3.5 years for the overall population;C.mongolicus:2 years for CRB, 4 years for LRB, and 3 years for the overall population) (Fish Research Laboratory, Hubei Institute of Hydrobiology, 1976; Xu et al., 2009;Yang, 2009). The substitution rate of 3% per million years for freshwater f ish species was generally applied in the CR (Donaldson and Wilson, 1999). The goodness-of-f it of the actual distributions were calculated by Harpending’s raggedness index (Hri)(Harpending, 1994) with the expected distributions using a model of population expansion, and the sum of squared deviations (SSD) by assessing signif icance with 1 000 permutations.
Third, Bayesian skyline plots (BSPs) (Drummond et al., 2005) were generated using BEAST 1.8.4 for CR with 20 million generations to explore demographic histories. This analysis was performed using the TrN substitution model forC.alburnusand the HKY substitution model forC.mongolicus, and no partition into codon positions. The coalescent tree prior was specif ied as the BSP. The uncorrelated lognormal relaxed-clock model, with means value(4±1.9 Ma) of the normal distributions was used. The remaining settings were as default.
3 RESULT
3.1 Sequence information
The mitochondrial CR successfully amplif ied 327C.alburnusfrom 11 populations and 223C.mongolicusfrom 9 populations.C.alburnussequences (GenBank Accession Nos. MG584736-MG584792) andC.mongolicussequences (GenBank Accession Nos.MG584793-MG584829) were deposited in GenBank(Table S1). ForC.alburnus, the aligned sequences(842 bp) contained 58 variable sites, of which 47 were parsimony-informative. ForC.mongolicus, the aligned sequences (932 bp) contained 51 variable sites, of which 49 were parsimony-informative. No insertions or deletions were detected in CR.
3.2 Culter alburnus phylogenetic analysis and divergence time estimate
Based onC.alburnusCR haplotypes, BI and ML methods yielded similar tree topologies and consistently supported two lineages (Fig.2a, only BI tree shown). Lineage I, the sister group of Lineage II,consisted of specimens from the ZRB. Lineage II included samples from the CRB, HRB, and LRB. The median-joining network analysis revealed a “weblike” topology pattern, whereas there were signif icant diff erences between the ZRB nodes versus the other three basin nodes (Fig.3a).
We conducted divergence time estimation among diff erent haplotypes ofC.alburnususing an uncorrelated lognormal relaxed-clock model. Based on the 95%credibility intervals (95% CI) of the posterior distribution (Fig.4a),C.alburnusdiverged from outgroups such asC.idellusin the Pliocene 5.25 Ma(95% CI: 4.18-6.79). The lineage I group diverged at the same series 2.74 Ma (95% CI: 1.96-3.48), and the other three basins appeared to share an obscure common ancestor and all diverged in the Pleistocene.
3.3 Culter alburnus genetic diversity and population structure
ForC.alburnus, the number of haplotypes (n),haplotype diversity (h), and nucleotide diversity (π)for each population are presented in Table 2. The overall CRh(0.913±0.008) and π (0.008 0±0.005 1)were high. A total of 57 haplotypes were detected from all sequences. The highest haplotype and nucleotide diversities were discovered in the Kuilei Lake (KLL) and Liangzi Lake (LZL) populations,whereas the lowest were discovered in the Weishan Lake (WSL) and STR. The haplotypes displayed restricted basin geographical distributions, with 98.2% of the haplotypes being unique for a single basin. Haplotype 7 (GenBank Accession No.MG584742) was the most common (shared by 64 individuals) and widespread, being shared among all CRB and HRB populations.

Fig.2 Bayesian inference analysis tree for Culter alburnus (a) and Culter mongolicus (b) based on CR haplotype
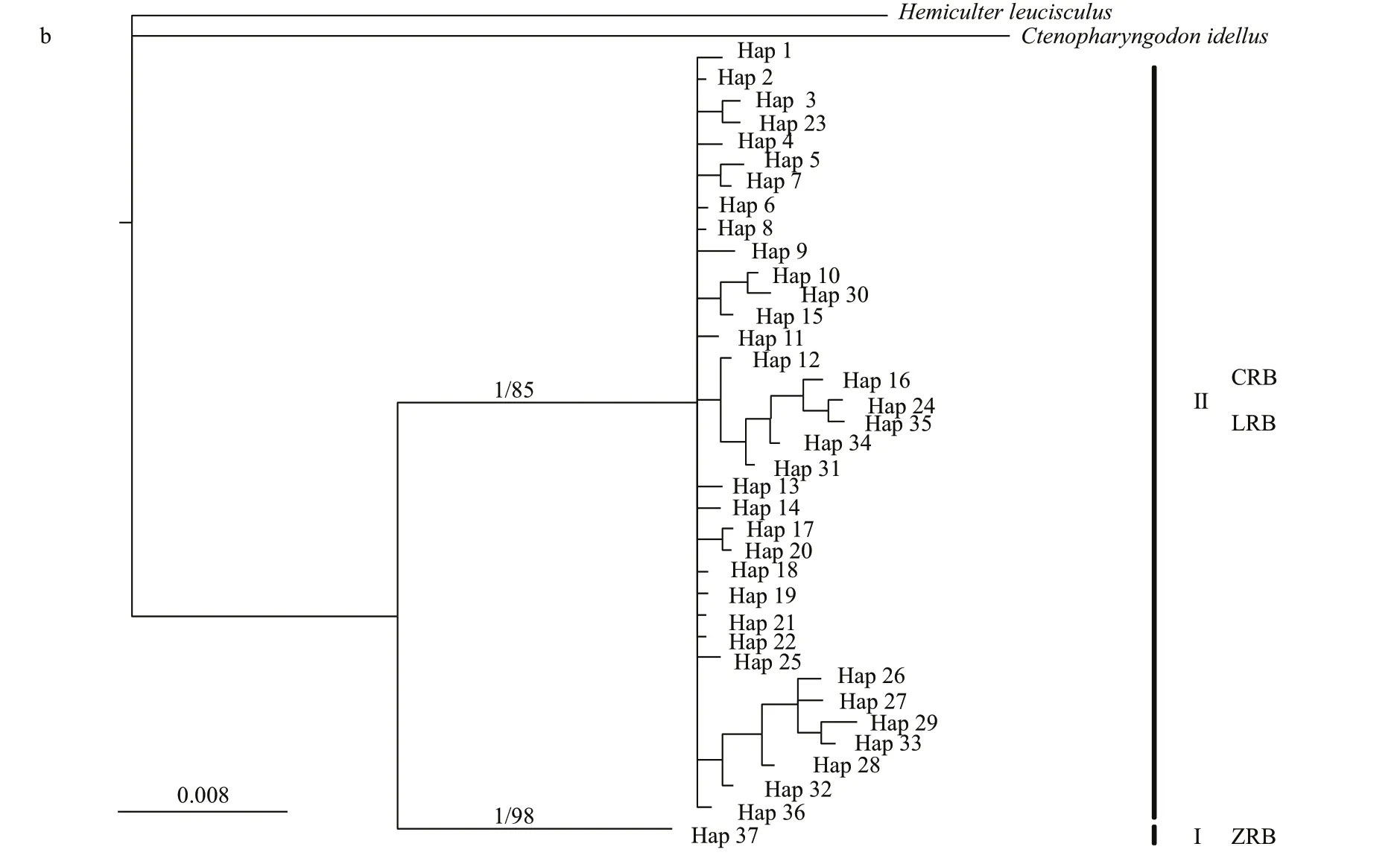
Fig.2 Continued
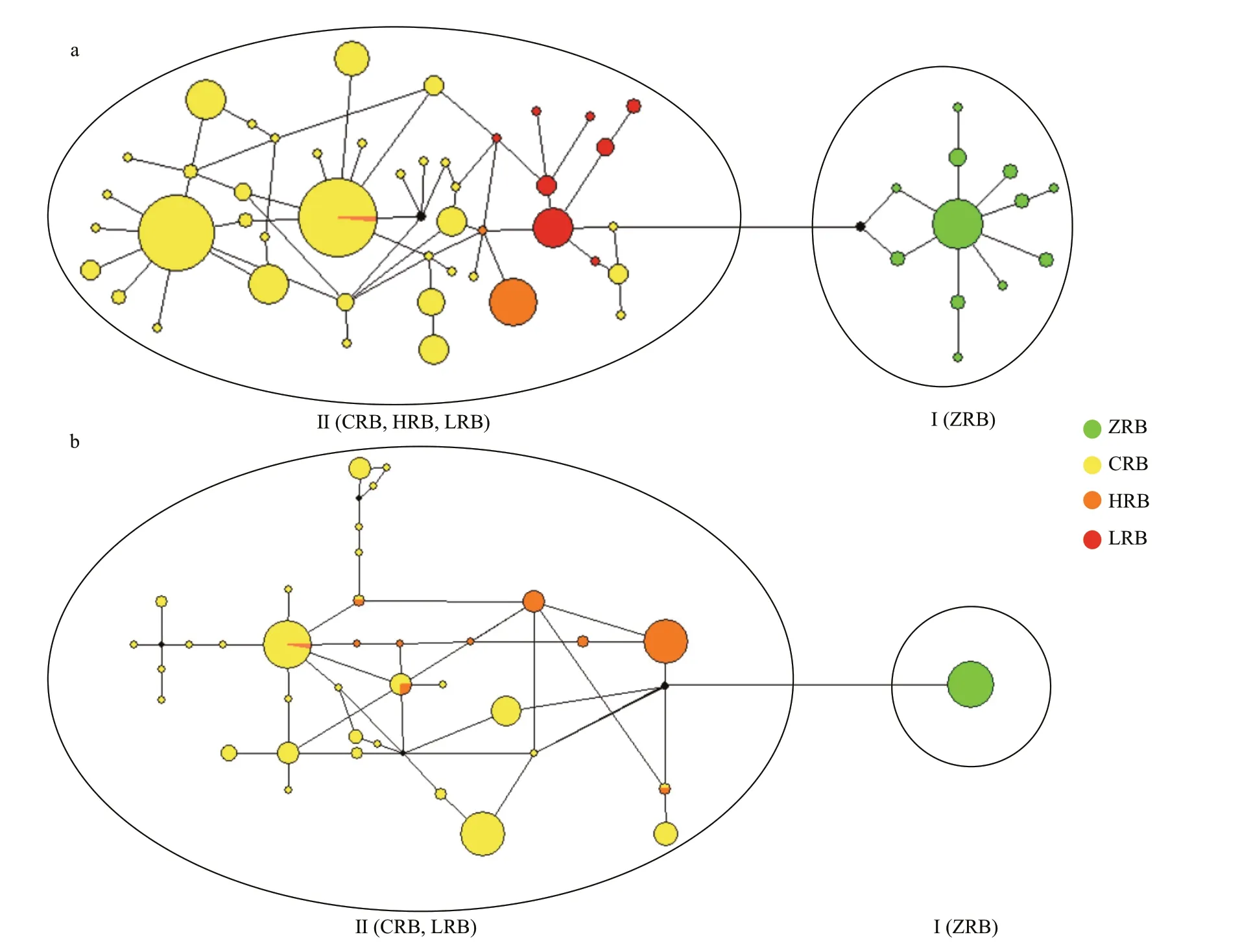
Fig.3 Median-joining network of haplotypes of Culter alburnus (a) and Culter mongolicus (b)
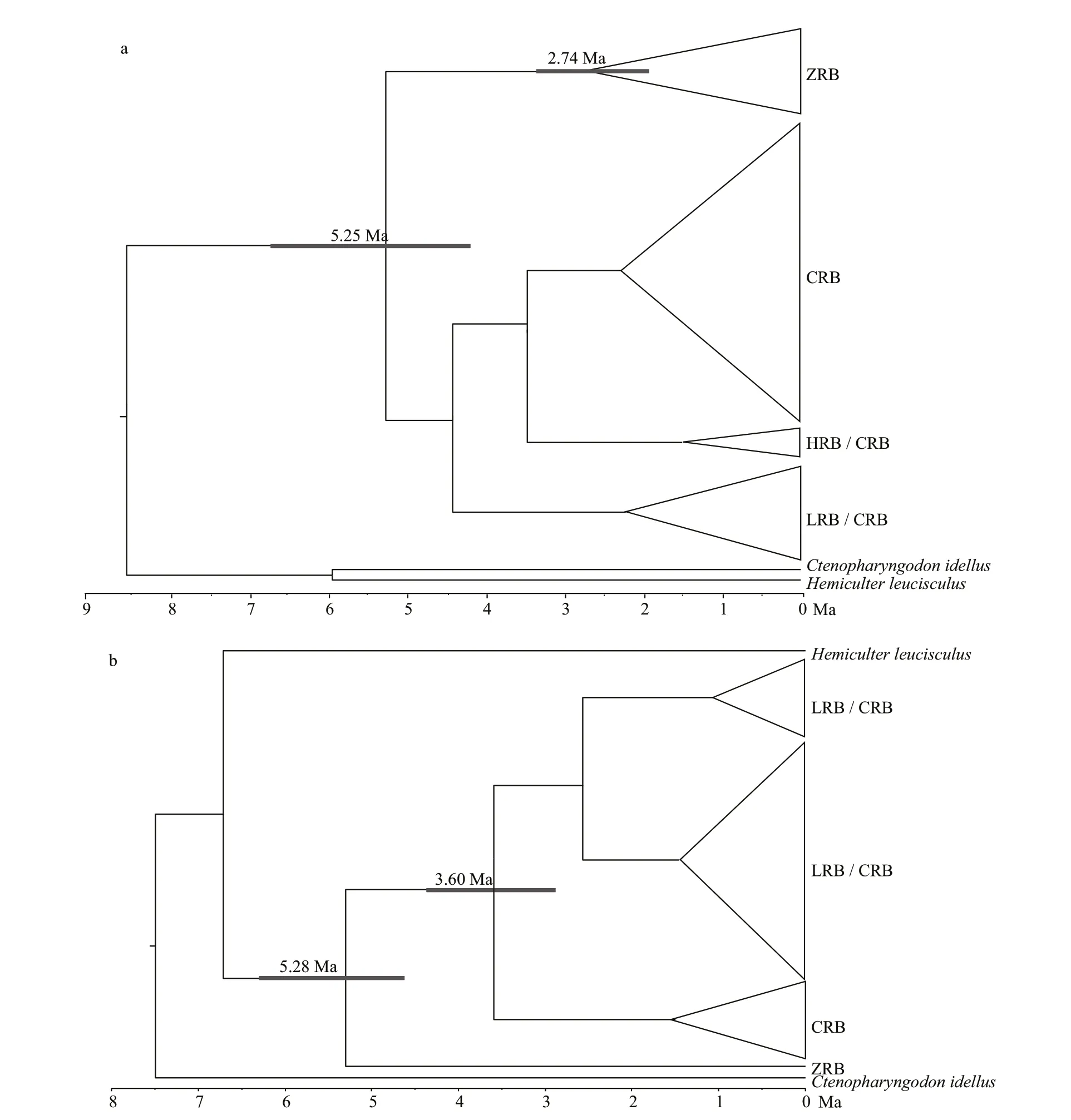
Fig.4 Divergence time tree for Culter alburnus (a) and Culter mongolicus (b)
Based on the K2P model, the mean genetic divergence within and between each populations are presented in Table 3. The overall average genetic divergence among individuals was 0.007 6 forC.alburnus. Among the 11 populations, the overall mean intrapopulation genetic distance of the LZL population (0.004 0) was largest and Xingkai Lake(XKL) (0.000 8) population was smallest. Genetic distances among CRB and HRB populations(0.002 1-0.005 6) were very small, but genetic distances between ZRB and other basin populations(0.020 2-0.021 7) were higher.
For population structure analysis, all samples were grouped by clades and basins. An AMOVA revealed that most of the variation (79.45%,P<0.000 1)occurred among clades (Table 4), whereas the variance among populations within groups (4.92%,P<0.000 1) and within populations (15.64%) was relatively small (FST=0.843 7,P<0.000 1), indicating a high level of clade structure. Pairwise comparisons of genetic diff erentiation (FST) values varied from-0.015 5 to 0.937 8. All pairwiseFSTcomparisons between basin populations were statistically signif icant. For CRB, signif icant genetic diff erences were observed between Shengjin Lake (SJL) and other CRB populations. The values ofNmcalculated from GammaSt (from 0.15 to 3.53) andFST(from0.07 to 0.45) indicated a limited gene f low level among the fourC.alburnusbasin populations,especially between the ZRB and the other three basin populations, which was in accordance with their signif icantFSTvalues (Table S2).
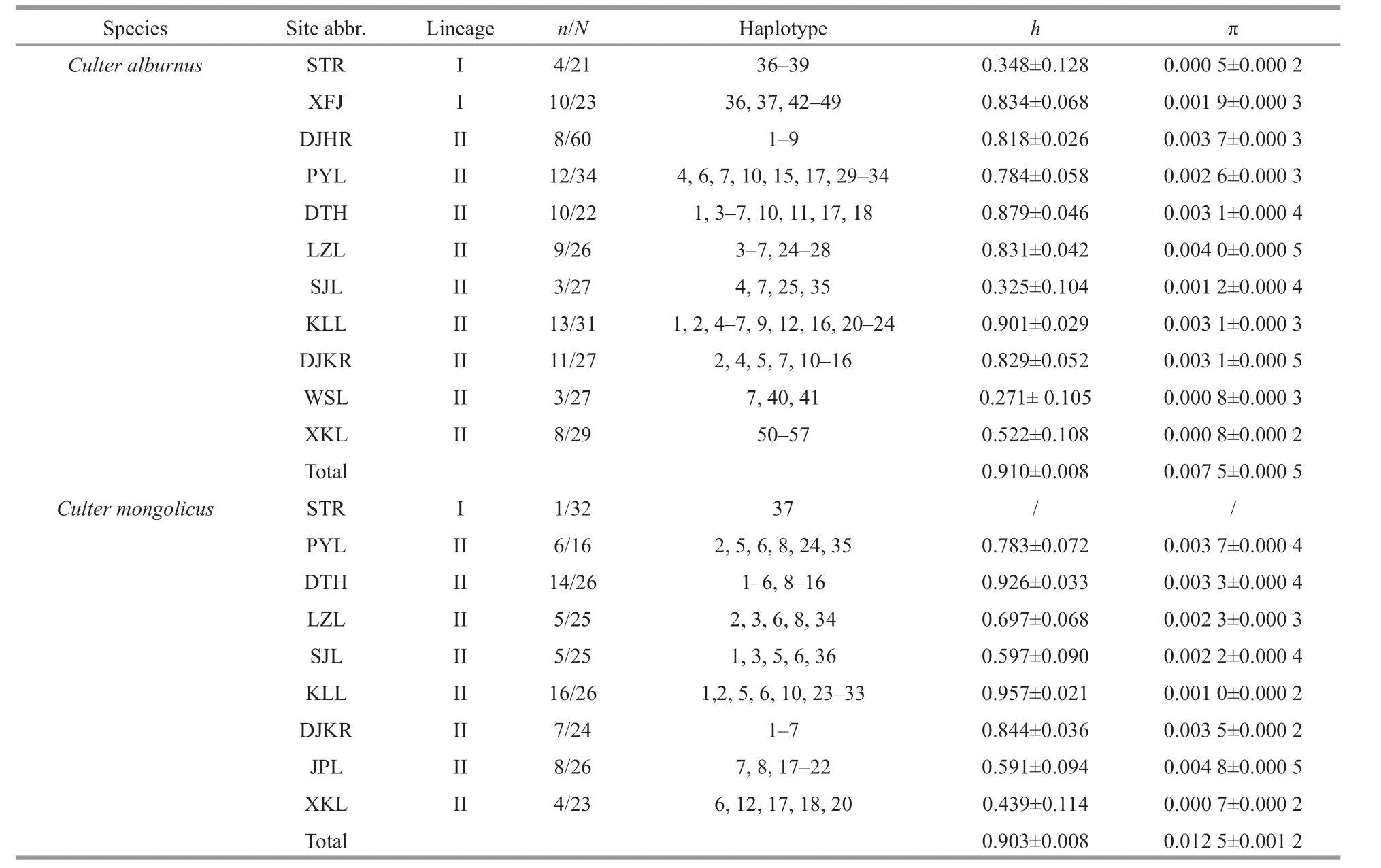
Table 2 Genetic diversity indexes for all Culter alburnus and Culter mongolicus populations
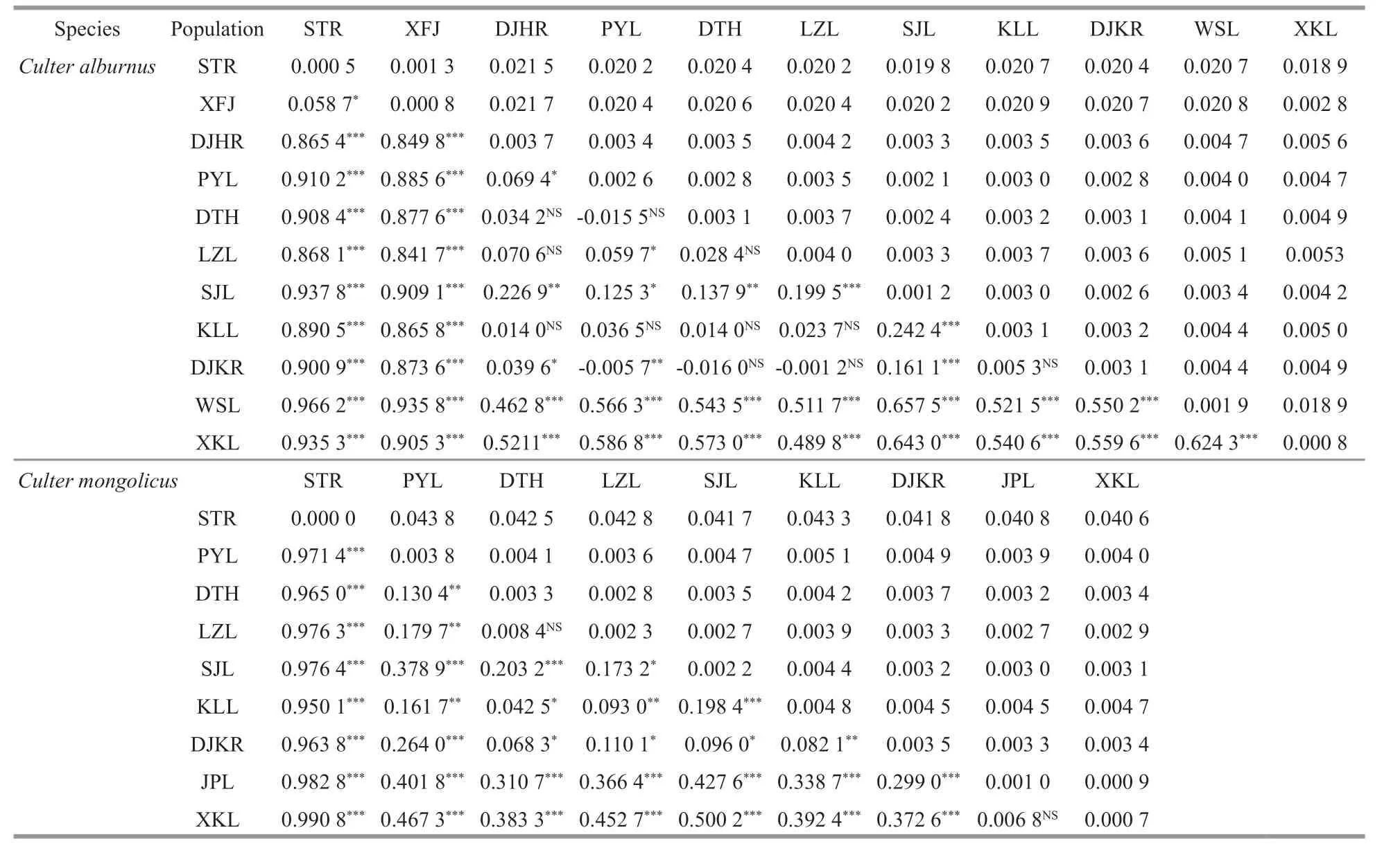
Table 3 Population pairwise F ST f ixation index and signif icant probability estimate (below the diagonal), mean genetic distances between (above the diagonal) and within (on the diagonal) populations of Culter alburnus and Culter mongolicus

Table 4 Analysis of molecular variance for Culter alburnus and Culter mongolicus populations
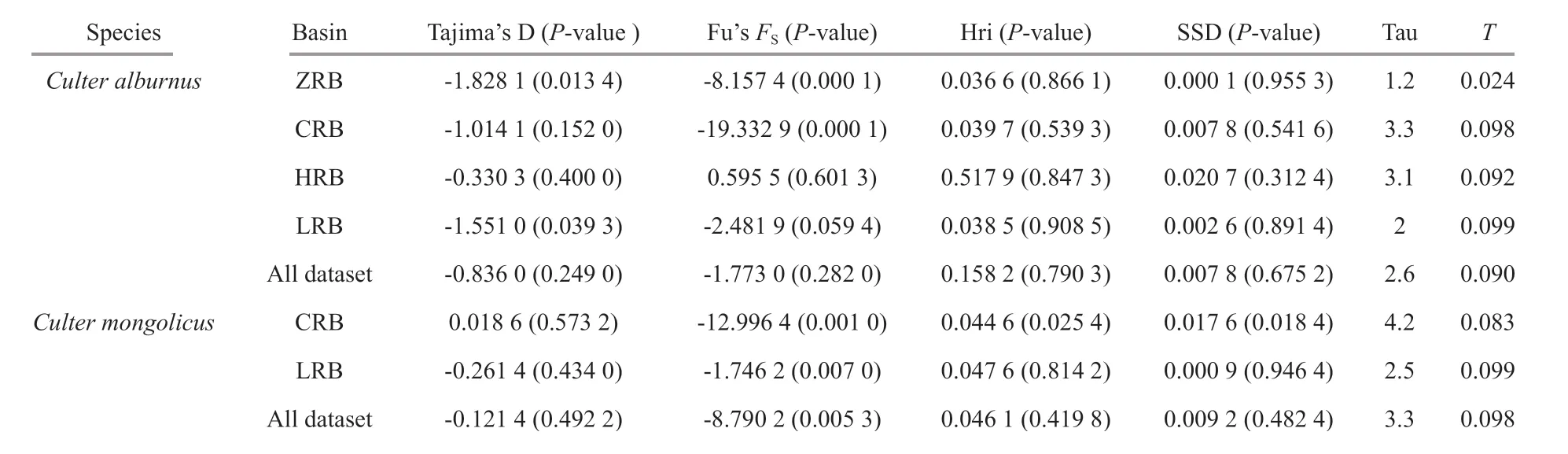
Table 5 Statistics of neutrality tests and mismatch distribution analysis for basin
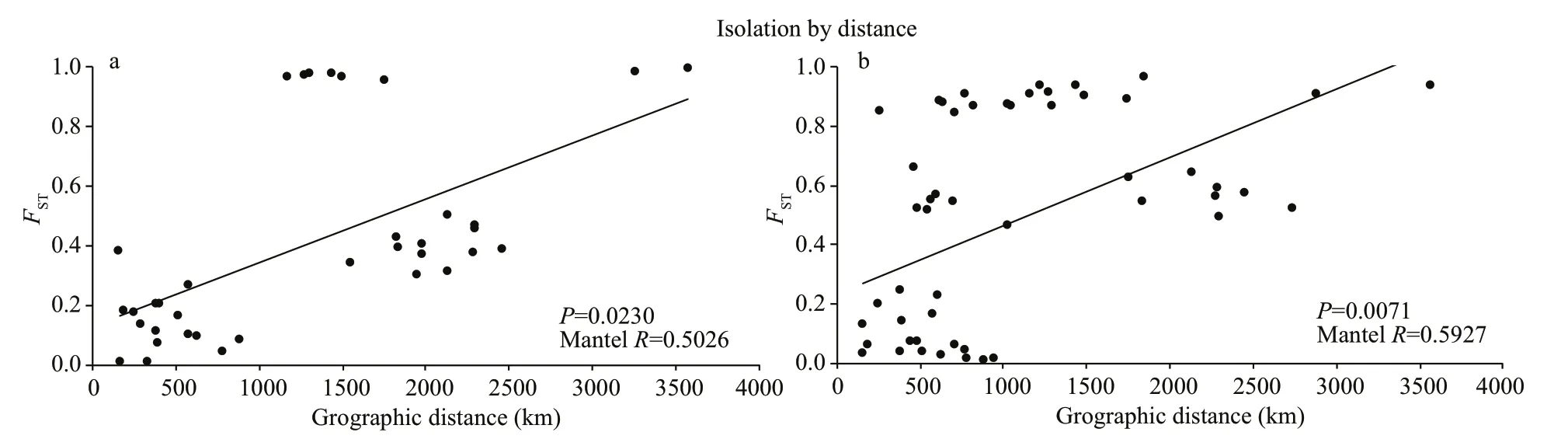
Fig.5 Correlation between genetic diff erences ( F ST) and geographic distances (km) of Culter alburnus (a) and Culter mongolicus (b)
There was a signif icant positive relationship between population genetic diff erentiation and geographic distance (Mantel tests,R=0.502 6,P=0.023 6<0.05) (Fig.5a).
3.4 Culter alburnus demographic history
ForC.alburnus, neutrality tests were negative except for the HRB, and Fu’sFStest values were statistically signif icant for ZRB, CRB, and LRB populations (P<0.05) (Table 5). Mismatch analysis revealed an approximately unimodal distribution of the overall population and ZRB, CRB, and LRB populations (Fig.6 left - a, b, c, e), but the shape of the mismatch distribution of HRB was approximately bimodal (Fig.6 left - d). Nevertheless, the model of sudden demographic expansion for the overall population was not rejected by the SSD and Hri values(Table 5). The total population ofτwas calculated as between 1.2 and 3.3 by Arlequin using the evolutionary rate calibrated in this study (3% per Ma), and the expansion time for each basin and overall was estimated to be between 0.035 and 0.098 Ma (Table 5).Additionally, BSP suggested that remarkable expansion occurred in the overall population and CRB and LRB populations (Fig.6 right - a, c, e), and that moderate expansion occurred in the ZRB population (Fig.6 right - b), but that the eff ective population size was relatively stable for the HRB population (Fig.6 right - d).
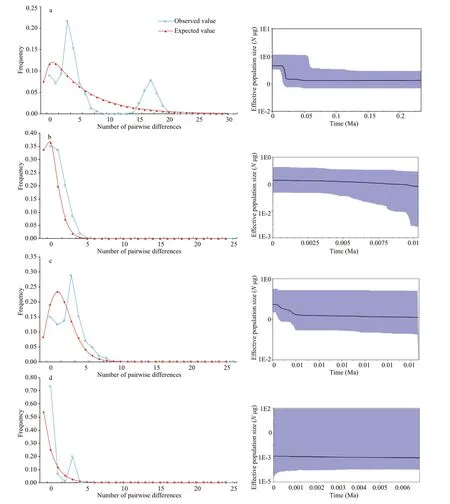
Fig.6 Mismatch distribution (left) and Bayesian skyline plot (BSP) analysis (right)
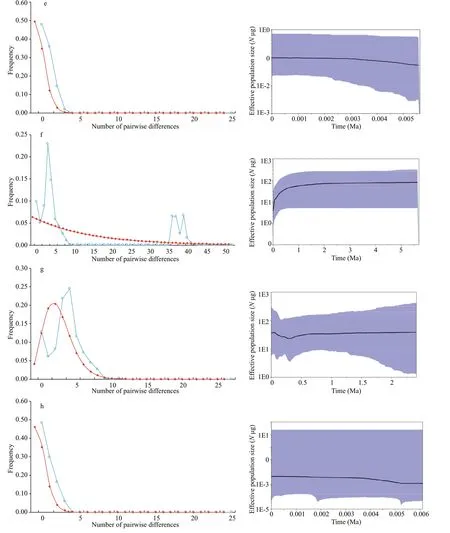
Fig.6 Continued
3.5 Culter mongolicus phylogenetic analysis and divergence time estimate
Based onC.mongolicusCR haplotypes, the BI and ML methods yielded similar topologies and consistently supported two lineages (Fig.2b, only the BI tree is shown). Lineage I, the sister group of Lineage II, consisted of specimens from the ZRB.Lineage II included samples from CRB and LRB. The median-joining network analysis revealed a “weblike” topology pattern, and someC.mongolicusindividuals from CRB and LRB population haplotypes shared the same node (Fig.3b). Specif ically,haplotypes 6, 7, 8, and 12 are shared by CRB and LRB populations, whereas signif icant diff erences between the ZRB nodes versus the other two basin nodes are seen.
Based on the 95% CI of the posterior distribution(Fig.4b),C.mongolicusdiverged from outgroups in the Pliocene 5.28 Ma (95% CI: 4.61-6.79 Ma), and lineage I group diverged at the same time; the lineage II basin appeared to share a common ancestor 3.60 Ma(95% CI: 2.84-4.43 Ma).
3.6 Culter mongolicus genetic diversity and population structure
ForC.mongolicus, the values ofn,h, π for each population are presented in Table 2. The overall CR h(0.903±0.008) and π (0.012 5±0.001 2) were high.Thirty-seven haplotypes were detected among all sites. The highest haplotype and nucleotide diversities were discovered in the KLL population. The haplotype displayed restricted basin geographical distribution,with 89.2% of the haplotypes being unique for a single basin, whereas there was only one haplotype(37, GenBank accession No. MG584829) for the ZRB(STR population).
The mean genetic divergence within and between each of the populations are shown in Table 3, and the overall average genetic divergence among individuals was 0.012 9 forC.mongolicus. Among the 9 populations, the overall mean intrapopulation genetic distance of the KLL population (0.004 8) was largest and the STR (0) population was smallest. The genetic distances among CRB and HRB populations(0.000 9-0.005 1) were very small, but those between the ZRB and other basin populations (0.040 6-0.043 8)were higher.
For population structure analysis, all samples were grouped by clades and basins. An AMOVA revealed that most of the variation (92.33%,P<0.000 1)occurred among groups (Table 4), whereas the variance within populations (5.53%,P<0.000 1) and among populations within groups (2.14%,P<0.000 1)was relatively small (FST=0.9447,P<0.000 1),indicating a high level of clade structure.
Pairwise comparisons of genetic diff erentiation(FST) values varied from 0.006 8 to 0.990 8. All pairwiseFSTcomparisons between basin populations were statistically signif icant. For the CRB, signif icant genetic diff erences were observed between CRB populations, with the exception of the DTH and LZL.The values ofNmcalculated from GammaSt (from 0.14 to 3.32) andFST(from 0.06 to 1.05) indicated a limited gene f low level amongC.mongolicusbasin populations, especially between the ZRB and the other two basin populations, which was in accordance with their signif icantFSTvalues (Table S2).
There was a signif icant positive relationship between population genetic diff erentiation and geographic distance (Mantel tests,R=0.592 7,P=0.007 1<0.05) (Fig.5b).
3.7 Culter mongolicus demographic history
ForC.mongolicus, the ZRB population was excluded from demographic analysis because of the unique haplotype. The neutrality test was negative for the overall population and the CRB and LRB. All Fu’sFStest values were signif icantly negative(P<0.01) (Table 5). The mismatch distribution shape ofC.mongolicuswas unimodal (Fig.6 left -f, g, h).Nevertheless, the model of sudden demographic expansion for the overall population and each basin population was not rejected by the SSD and Hri values (Table 5). The total populations ofτwere calculated between 2.5 and 4.2 using Arlequin, and the expansion times for each basin and the overall population were estimated to be between 0.083 and 0.099 Ma (Table 5). Similar results were obtained with BSP, suggesting that moderate expansion occurred in the overall population and the CRB and LRB (Fig.6 right - f, g, h).
4 DISCUSSION
4.1 Population divergence and implication of cryptic subspecies
Phylogenetic analysis based on CR revealed two lineages (I and II), which is highly consistent with the geographical patterns forC.alburnusandC.mongolicus(Fig.2). The divergence time estimate revealed a deep divergence analogous to phylogenetic analysis. Lineage I was the f irst to split from Lineage II in the Pliocene and exhibits a high divergence from it; Lineage II diverged during the Pleistocene but at a later point in time. Lineage I and populations from Guangdong Province and Hainan Province were found to show a close relationship, though they are separated by the Qiongzhou Strait. However, this may be because the Qiongzhou Strait was once part of the coastal plain of the China mainland continent during the Pleistocene glaciations (Wang et al., 2012).
There is a deep genetic divergence between ZRB populations and other populations (C.alburnus,0.020 2-0.021 7;C.mongolicus, 0.040 6-0.043 8;Table 3) based on genetic distance calculations. This level of divergence has previously been attributed to subspecies-level diff erences (Shaklee et al., 1982).Therefore, we suggest thatC.alburnusandC.mongolicusof ZRB populations represent cryptic subspecies. BI tree analysis and web-like statistical haplotype network f indings also support this. The signif icant pairwise genetic diff erentiation detected between the ZRB populations and other populations is in accordance with that of many Cyprinidae species in China such asAcheilognathusmacropterus(Zhu and Liu, 2007),C.idellus(Fu et al., 2015),Hypophthalmichthysmolitrix(Ji et al., 2009), andH.leucisculus(Chen et al., 2017). It is conceivable that geographical isolation is a key factor in population diff erentiation. For example, the ZRB was isolated from the CRB by the Nanling Mountains. Moreover,compare to the geographic distance between the two ZRB populations (XFJ and STR) is 710 km, the distance between the DJHR (CRB) and XFJ (ZRB) is only 260 km, but the two basin populations show completely diff erent haplotypes. Gene f low has also been detected among the basin populations.Additionally, there is evidence of limited gene f low between the ZRB and other basin populations.Second, historical factors have played an important role in determining the patterns of genetic variability.ExtantCulteroriginated from the Pliocene, and the ZRB was also diverged during the late Pliocene period (Wang et al., 2012). Therefore, the ZRB populations were isolated from their point of origin at an early stage ofCulterdivergence. Divergence time estimations support this hypothesis. Third, we consider that ZRB belong to peripheral area and play a role in genetic diff erentiation. Moreover,Culterspecies are a temperate f ish (Froese and Pauly, 2017)but the ZRB is a tropical river (Dudgeon, 2000), and peripheral populations tend to occur in more unsuitable environments than central populations(Lesica and Allendorf, 1995).
4.2 Species-level of genetic diversity and structure
Based on haplotype and nucleotide diversities,C.alburnusandC.mongolicusboth showed high overall genetic diversities as seen in previous f indings(Wang et al., 2007; Qi et al., 2013, 2015; Yang et al.,2016). This could ref lect the complex and variable heterogenetic habitat ofCulterpopulations, which are widely distributed in China. However, the genetic diversity within populations shows that some populations have high levels of diversity, while others have low levels. All CRB populations typically show higher genetic variability than the other three basin populations forC.alburnus, whereas a similar level of variability was found among theC.mongolicuspopulations. Genetic patterns and species diversity may have been aff ected by speciation events and subsequent radiations (Xiao et al., 2009; Yamaguchi et al., 2010). Additionally, diff erences in the time of species origins have contributed to the general positive correlation with genetic diversity (Messmer et al., 2012). Populations near the center of a species′range are usually contiguous and at a high density,while populations at the margins tend to be isolated and sparse. Thus, peripheral populations are small and isolated, with low genetic variability (Brussard,1984; Lesica and Allendorf, 1995). We therefore deduced that CRB populations were at or near the core region of their distribution. This result was also supported by the fact that the RPEC was the place of origin ofCulterspecies (Liu and Su, 1962; Chen,1998; Wang, 2013).
Other notable discoveries include the low genetic diversity of SJL and Dangjiangkou Reservoir (DJKR)population and the signif icant genetic diff erentiation between SJL and other CRB populations. This result is mainly caused by geographic isolation. The SJL sluice and DJKR reservoir were constructed between 1950-1970, and were isolated from the main stream of the Changjiang River. Major threats to SJL include artif icial reclamation, serious sedimentation accumulation, and habitat degradation (Jiang et al.,2007). The DJKR dam prevents f ish from moving through, thereby inducing geographic isolation between the DJKR and the Changjiang River. These factors can aff ect genetic diversity by decreasing gene f low among the CRB.
The main aim of population genetic studies is to evaluate levels of genetic connectivity among populations. Major factors that shape genetic structure among populations include population size, dispersal potential, environmental conditions, and life history strategies (Gaggiotti et al., 2009). In the present study,AMOVA and pairwiseFSTvalues indicated that high levels of population structure and genetic diff erentiation occurred in diff erent drainage basin populations forC.alburnusandC.mongolicus. The fact that 98.2% and 89.2% of haplotypes were only detected in singleC.alburnusandC.mongolicusbasin localities, respectively, indicates that limited gene f low occurred among drainage basin populations.
Allopatric speciation has a central role in generating new species by reproductive isolation(April et al., 2013). Freshwater f ish populations are usually conf ined to conf ined areas during a relatively short to moderate evolutionary period (Dewoody and Avise, 2000; Yamaguchi et al., 2010). During the Pleistocene glacial period, temperate freshwater f ish experienced landscape changes s in intermittent connected and isolated waterways and glacial refugia,leading to complex phylogeography and many opportunities to study recent or incomplete speciation(Behnke, 1972; Bernatchez and Wilson, 1998; Taylor,1999; Ruzzante, 2011; Ruskey and Taylor, 2016).The clear pattern of IBD inC.alburnusandC.mongolicusin the current study supports the hypothesis and was predicted by the drainage basin isolation. Geographic distance could be a reasonable explanation for the observed geographic distribution of genetic variation between populations. The two largest carnivorousCulterspecies mainly coexist in larger rivers, lakes, and reservoirs in China. Perhaps because of the demand for food resources they rarely inhabit small tributaries, and gene f low among basins may be rare too. Moreover, becauseC.alburnusandC.mongolicusspawn in f lowing water (Fish Research Laboratory, Hubei Institute of Hydrobiology, 1976)and are potamodromous, gene f low among populations within the same drainage basin may be more common.
4.3 Demographic process of Culter
Climatic oscillation played an important role in the contemporary diversity of many species and communities. In particular, climate changes between ice age maxima and warm interglacials caused large changes in the distribution of species, which ultimately aff ect the distribution of genetic variation within species (Hewitt, 2003). During the Pleistocene,f ive glaciations occurred in Eastern China (Yi et al.,2007). The last interglacial period occurred at approximately 0.1 Ma, while glacial retreat occurred at 0.075 Ma. The last glaciation (LG) in Eastern China started at approximately 0.073 Ma and continued until 0.016 Ma, and the last glacial maximum (LGM) development in Eastern China occurred during the 0.024-0.016 Ma.
On the basis of estimation expansion times from mismatch distribution, mostCulterpopulations(C.alburnus, 0.035-0.098 Ma;C.mongolicus: 0.083-0.099 Ma) underwent a period of population expansion that began before the LGM in Eastern China. This is related to suitable environments within their areas during interglacial periods (0.1-0.075 Ma). Typical features of this type of expansion pattern are supported by high haplotype diversity but low nucleotide diversity, signif icantly negativeFSvalues, unimodal distribution of pairwise diff erences, and expansion BSP analysis (Nei, 1987; Tajima, 1989; Rogers and Harpending, 1992; Fu, 1997; Drummond et al., 2005).
However, BSP and Tajima’s analysis suggest that eff ective population size was relatively low and stable for the HRBC.alburnuspopulation. Two reasons account for this abnormal phenomenon. First,according to divergence time estimations, the HRB was the latest diverged group (1.48 Ma) forC.alburnus. Furthermore, its genetic diversity was lowest inC.alburnus. Since the Pleistocene ice age,the population bottlenecks appear to have impacted the extent of haplotype diversity in many freshwater f ish populations (Billington and Hebert, 1991) and were inf luenced by the small founder eff ect later(Hoff man and Blouin, 2004). Second, habitats of the HRB (WSL, 34°34′ 45″N), which were distributed in the southern boundary of the permafrost area in the hilly plain region of Eastern China (near 33°20′ N-33°40′ N) (Pu, 1991), were unsuitable forC.alburnusduring the LG period because they are temperate f ish.
4.4 Conservation implication
CulteralburnusandC.mongolicusare economically valuable freshwater f ish that are widely distributed in larger rivers, lakes, and reservoirs of China. However, because of their carnivorous nature,they were once considered harmful and were removed from water bodies. Today, some anthropogenic activities such as illegal f ishing, random stock enhancement, and habitat degradation (e.g., dam construction and water pollution) are major threats to these species and result in the miniaturization of natural f ish resources. Indeed, during the last few decades, the wild populations have declined so rapidly(Liu et al., 2007) that conservation of their genetic diversity has become a very important issue.
Maintaining genetic diversity is a fundamental concern in conservation and evolutionary biology,and is an important part of biodiversity. Populations lacking genetic variability have low adaptability to stress, while populations with high genetic variability can rapidly respond to environmental changes (Ayala,1965; Nei et al., 1975; Frankham et al., 2002). We propose several recommendations based on the current study. Each basin (ZRB, CRB, HRB, and LRB) should be considered an individual Management Unit based on the signif icant genetic divergence occurring in diff erent drainage basin populations.Additionally, the CRB population demands priority protection because it has the highest genetic diversity and provides the largest yield ofCulterspecies, thus we should protect the integrity and continuity of their habitats and reduce human disturbance. Special attention should also be paid to SJL and DJKR populations because dam construction and artif icial reclamation will lead to geographic separation and impede gene f low among basins. The most important course of action should be to reconnect the lakes with the Changjiang River, this is especially important for the restoration of genetic diversity in smaller lakes,thus we recommend installation f ish passage facilities for DJKR and restoration of the connectivity with Changjiang River for SJL. TheC.mongolicuspopulation of the ZRB (STR) also requires a great deal of attention because of its genetic uniqueness,which includes exclusive mtDNA CR haplotypes.Long-term monitoring programs should also be established. Finally, to understand genetic patterns and establish eff ective conservation strategies for the twoCulterspecies, more sensitive nuclear markers and larger samples should be employed in future research.
5 CONCLUSION
This study assembled a relatively larger scale dataset to inferCulterspecies phylogeographic patterns in China. The results support the hypothesis that geographic isolation drove divergence between lineages I and II. Based on the molecular clock,Culterspecies appeared to originate during the early Pliocene. Genetic diversity diff erences among basin populations are signif icant forCulterspecies, and demographic analysis indicated that most populations experienced population expansion during the warm interglacial period. Based on the current study, we propose several suggestions for the protection of genetic diversity: each basin should be considered an individual Management Unit and CRB populations demand priority protection; theC.mongolicusSTR population possesses unique genotypes, which calls for a special conservation concern for this species at the population level.
6 DATA AVAILABILITY STATEMENT
Sequence data that support the f indings of this study have been deposited in GenBank under accession numbers from MG584736 to MG584792 forC.alburnusand from MG584793 to MG584829 forC.mongolicus. Details regarding individual samples are available in Table S1.
7 ACKNOWLEDGMENT
We would like to acknowledge colleagues at the Fisheries Ecology Lab for their assistance in f ield sampling.
 Journal of Oceanology and Limnology2020年2期
Journal of Oceanology and Limnology2020年2期
- Journal of Oceanology and Limnology的其它文章
- Contribution of surface wave-induced vertical mixing to heat content in global upper ocean*
- Upper ocean response to typhoon Kujira (2015) in the South China Sea by multiple means of observation*
- Inf luence of simulating deep-sea environmental factors on cathodic performance of seawater battery*
- Adsorption characteristics of chitooligosaccharides onto activated charcoal in aqueous solutions*
- Eff ects of hypoxia on survival, behavior, and metabolism of Zhikong scallop Chlamys farreri Jones et Preston 1904*
- Distinct inf luence of trimethylamine N-oxide and high hydrostatic pressure on community structure and culturable deep-sea bacteria*