
BE3型胞嘧啶碱基编辑器在谷氨酸棒杆菌中的开发及应用
2020-03-19 09:32黄华媚白立宽刘叶李俊维王猛花尔并
生物技术通报 2020年3期
黄华媚 白立宽 刘叶 李俊维 王猛 花尔并
(1. 天津科技大学生物工程学院,天津 300457;2. 中国科学院系统微生物工程重点实验室,天津 300308;3. 中国科学院天津工业生物技术研究所,天津 300308)
David Liu研究团队首次报道了基于鼠源胞嘧啶脱氨酶(rat APOBEC1,rAPOBEC1)与d/nCas9蛋白融合的胞嘧啶碱基编辑器BE(Base editor),实现在哺乳动物细胞中胞嘧啶(Cytosine,C)到胸腺嘧啶(Thymine,T)的单碱基转换[4]。经过优化,融合rAPOBEC1、nCas9(D10A)以及尿嘧啶糖苷酶抑制蛋白(Uracil DNA glycosylase inhibitor,UGI)的BE3型胞嘧啶碱基器(rAPOBEC1-XTEN-nCas9(D10A)-UGI)编辑效率得到较大幅度提高,并在其它动植物和微生物中广泛应用。与此同时,Akihiko Kondo研究团队则将七鳃鳗来源的胞嘧啶脱氨酶(PmCDA1)与d/nCas9蛋白进行结合,开发了胞嘧啶碱基编辑器Target-AID(Activation-induced cytidine deaminase),实现在酵母和哺乳动物细胞中的应用[5]。随后,David Liu研究团队又突破性的开发了基于腺苷脱氨酶与CRISPR/Cas系统相结合的腺嘌呤碱基编辑器,可实现腺嘌呤(Adenine,A)到鸟嘌呤(Guanine,G)的单碱基转换,进一步的丰富了碱基编辑系统的工具箱[6]。
谷氨酸棒杆菌(Corynebacterium glutamicum)是一株生物安全且具有重要工业应用价值的底盘微生物,目前被广泛应用于各种大宗化学品的工业化生产中[7]。本研究团队率先在谷氨酸棒杆菌中开发了一种多元自动化的胞嘧啶碱基编辑方法(Multiplex automatedCorynebacterium glutamicumbase editing method,MACBETH),结合 PmCDA1与 nCas9(D10A)蛋白,实现C到T的编辑[8];随后,通过采用识别不同PAM的Cas9突变体,优化向导RNA(guide RNA,gRNA)的设计,开发腺嘌呤碱基编辑器等,进一步丰富了碱基编辑系统在谷氨酸棒杆菌中的应用[9]。而截至目前,应用较为广泛的、基于BE3结构的胞嘧啶碱基编辑器尚未有在谷氨酸棒杆菌中的报道应用。由于BE3碱基编辑器的编辑窗口(PAM序列上游-13到-17位)与现有MACBETH技术编辑窗口(PAM序列上游-16到-20位)存在明显差异,BE3型胞嘧啶碱基编辑器在谷氨酸棒杆菌中的开发与应用将进一步丰富基因组可编辑位点数量,为谷氨酸棒杆菌的工业化改造提供更多的选择。
1 材料与方法
1.1 材料
1.1.1 菌种和质粒 本研究所用菌株和质粒均为作者所在实验室保存和构建,详见表1。

表1 本文所用的质粒与菌株

表2 本文使用到的引物
1.1.2 酶、引物及相关试剂盒 Q5 High-Fidelity DNA聚合酶、T4 DNA连接酶、各种限制性内切酶购自NEB公司。大肠杆菌感受态细胞制备试剂盒购自TaKaRa公司。2×Es Taq Master Mix(Dye)购自康为世纪有限公司。质粒小量抽提试剂盒购自天根生化科技有限公司。重组试剂盒ClonExpress®MultiS One Step Cloning Kit购自南京诺唯赞生物科技有限公司。DNA回收试剂盒EZ geneTMGel/PCR Extraction Kit购自Biomiga公司。PCR引物由擎科生物有限公司合成。
1.1.3 培养基 LB培养基(g/L):蛋白胨10 g,氯化钠10 g,酵母粉5 g;NCM培养基(g/L):磷酸氢二钾17.4 g,氯化钠11.6 g,葡萄糖5 g,蛋白胨5 g,酵母粉1 g,柠檬酸三钠0.3 g,硫酸镁0.05 g,山梨醇 91 g,调 pH 至 7.2;BHIS 培养基(g/L):BHI 18.5 g,山梨醇91 g,调pH至7.2;LBHIS 培养基(g/L):蛋白胨5 g,氯化钠10 g,酵母粉2.5 g,BHI18.5 g,山梨醇91 g。制备固体培养基时添加2%的琼脂。
1.2 方法
1.2.1 gRNA的设计 本研究所选用的gRNA序列均由sgRNAcas9软件设计得出[10],设计参数为:gRNA长度20 nt,GC含量30%-70%,采用DNA双链计算,其他参数为软件默认参数。
1.2.2 质粒构建 质粒pXMJ19-rACTS的构建:首先将鼠源胞嘧啶脱氨酶rAPOBEC1的序列按照谷氨酸棒杆菌密码子的偏好性进行密码子优化,由金斯瑞公司合成;以合成质粒为模板,用引物对rAPF/lk-R扩增获得rAPOBEC1序列片段;以实验室现有质粒pTadA-TadA7.10-nCas9(D10A)TS为模板,用引物对nC9-F/CAG-R2、CAG-F3/9sgcx-R和9sgcx-F/SD-R分别扩增得到另外3个载体框架片段;将上述获得的四片段同源重组连接,具体操作参见ClonExpress® MultiS One Step Cloning Kit说明书,然后转化E. coliDH5α感受态;最后,经PstI和NdeI双酶切验证,并对质粒进行测序,获得正确质粒。
质粒pXMJ19-rACUTS的构建:将枯草芽孢杆菌(Bacillus subtilis)噬菌体PBS1来源的UGI蛋白的编码基因进行谷氨酸棒杆菌密码子优化,并由金斯瑞公司合成;以合成质粒为模板,用引物对UGI-F/R扩增得到UGI片段;以质粒pXMJ19-rACTS为模板,扩增获得其他载体片段;通过多片段同源重组、转化E. coliDH5α感受态、酶切验证以及测序,最终获得正确质粒。
定理1 (Rolle中值定理)若f满足:(i)f在闭区间[a,b]上连续;(ii)f在开区间(a,b)内可导;(iii)f(a)=f(b),则在(a,b)内至少存在一点ξ,使得f′(ξ)=0.
质粒pXMJ19-rACU-gRNA::ccdBTS的构建:以实验室保存质粒pnCas9(D10A)-AID-gRNA-ccdBTS为模板,用引物CAG-gRNA-F/SD-R、CAGF3/180824-R分别扩增得到两个含ccdB基因的骨架片段;以pXMJ19-rACUTS为模板,用引物rAPF/cx180322-2R、cx180322-2/UGI(-BsaI)-R、UGI(-BsaI)-F/CAG-R2分别扩增得到含rAPOBEC1-nCas9和UGI编码基因的片段;多片段同源重组后,转化到E. coliDB3.1感受态;经酶切验证及测序,最终获得正确质粒。
含不同靶标位点gRNA序列的质粒pXMJ19-rACU-gRNATS,均由质粒pXMJ19-rACU -gRNA::ccdBTS通过Golden Gate方法构建,具体方法可参见文献[8]。
1.2.3 谷氨酸棒杆菌的转化 将在BHI培养基中过夜培养的C. glutamicumATCC 13032菌液以初始OD6000.3转接到含INH、Tween80、DL-苏氨酸和葡萄糖的NCM培养基中。当OD600=1时,制备C.glutamicumATCC 13032感受态细胞感受态,具体方法参见文献报道[11]。
双质粒转化:取 2 μg pXMJ19-rACTS质粒或pXMJ19-rACUTS质粒和 1 μg pTrcmob-gRNA 加入到80 μLC. glutamicumATCC 13032感受态细胞中,1.8kV电转后立即加入1 mL 提前预热的BHIS培养基,放至金属浴锅46℃温浴6 min。摇床30℃培养2 h后,涂布于含15 μg/mL Kan和5 μg/mL Cm抗性的LBHIS固体培养基平板上,30℃静置培养2-3 d,直至长出单菌落。
单质粒转化:取1 μg pXMJ19-rACU-gRNATS质粒到80 μLC. glutamicumATCC 13032感受态细胞中,转化方法同双质粒,30℃后培养1 h后,涂布于含5 μg/mL Cm抗性的LBHIS固体培养基平板上。
1.2.4 谷氨酸棒杆菌的培养及碱基编辑 种子培养:挑取生长良好的单菌落接种于装有3 mL LBHIS液体培养基(含15 μg/mL Kan和5 μg/mL Cm)的15 mL试管中,30℃,220 r/min培养24 h。诱导培养:将种子培养液按初始OD6000.2的接种量转接于装有3 mL的LBHIS液体培养基(含1 mmol/L IPTG,15μg/mL Kan和5 μg/mL Cm)的15 mL试管中,30℃,220 r/min培养20 h。1.2.5 质粒去除及碱基编辑比例分析 将诱导编辑后的菌液,4 500 r/min、4℃条件下离心5 min,收集菌体,将上清去除后,用生理盐水(0.9% NaCl)将菌体洗两次,去除残留的IPTG,然后用等体积的生理盐水重悬菌体;按初始OD6000.01的接种量转接至不含抗性的LBHIS液体培养基中,37℃,220 r/min培养24 h;将菌液划线到无抗的LBHIS固体平板上,37℃静置培养2-3 d,直至长出单菌落;针对不同的靶标位点,随机各自挑取10个单菌落,用各自引物(距离靶标位点上下游200 bp处设计引物)和2×Es Taq Master Mix(Dye)进行菌落PCR,将PCR产物送Sanger测序。借助SnapGene软件分析Sanger测序峰图结果,并通过DNAMAN软件进行多序列比对分析,将编辑后单菌落测序结果与原始序列比对,汇总整理编辑效率、编辑位点及比例。
2 结果
2.1 基于rAPOBEC1-nCas9(D10A)融合蛋白的碱基编辑器的构建与测试
为了测试rAPOBEC1-nCas9(D10A)融合蛋白是否可以实现在谷氨酸棒杆菌中的高效编辑,我们以改造过的温敏型质粒pXMJ19TS作为框架,选用IPTG诱导型启动子tac,将经过谷氨酸棒杆菌密码子优化后的rAPOBEC1融合在nCas9(D10A)蛋白的N端,并在中间添加XTEN连接肽,获得重组质粒pXMJ19-rACTS(图1-A)。选取调控因子Cgl0016及Cgl0170作为两个靶标基因,将实验室前期已构建且含该两个靶标基因gRNA序列的质粒pTrcmob-gRNANoPer,分别与pXMJ19-rACTS共转化至C. glutamicumATCC 13032中。经过诱导编辑以及Sanger测序分析,如图1-B所示,两个位点均发生了编辑,但只有Cgl0170出现C到T的编辑,编辑比例仅为20%,而且两个位点均出现C到A或C到G的编辑,编辑产物纯度不高,推测原因可能为胞内的尿嘧啶DNA糖基化酶(Uracil DNA glycosylase,UDG)能够将中间产物U切除,形成无嘧啶位点,经过跨损伤合成聚合酶作用及DNA复制等,以一定几率将C碱基转换为其他碱基[12]。
2.2 融合UGI蛋白,构建并测试BE3碱基编辑器
在之前的研究报道中,尿嘧啶糖苷酶抑制蛋白UGI的添加能够有效抑制体内的DNA碱基切除修复机制,防止胞嘧啶核苷脱氨转化生成的尿嘧啶核苷被切除,从而提高C到T的转化效率,并且对提高编辑产物纯度也有重要作用[12]。为提高碱基编辑器的编辑效率,我们在上述rAPOBEC1-nCas9(D10A)融合蛋白基础上,在其C端进一步融合谷氨酸棒杆菌密码子优化后的UGI蛋白,获得BE3型胞嘧啶碱基编辑器(rAPOBEC1-XTEN-nCas9(D10A)-UGI),并将构建的质粒命名为pXMJ19-rACUTS(图2-A)。经过对靶标基因Cgl0016及Cgl0170的诱导编辑以及Sanger测序分析,相比较于rAPOBEC1-nCas9(D10A)融合蛋白,添加UGI后,两个位点的编辑效率均得到明显提高(图2-B)。其中,Cgl0016中C到T的编辑比例由原来的未编辑提高到高达90%,Cgl0170中C到T的编辑比例也由20%提高到70%,并且两个位点的编辑中均未再出现C到A,C到G的编辑,编辑产物纯度提高。
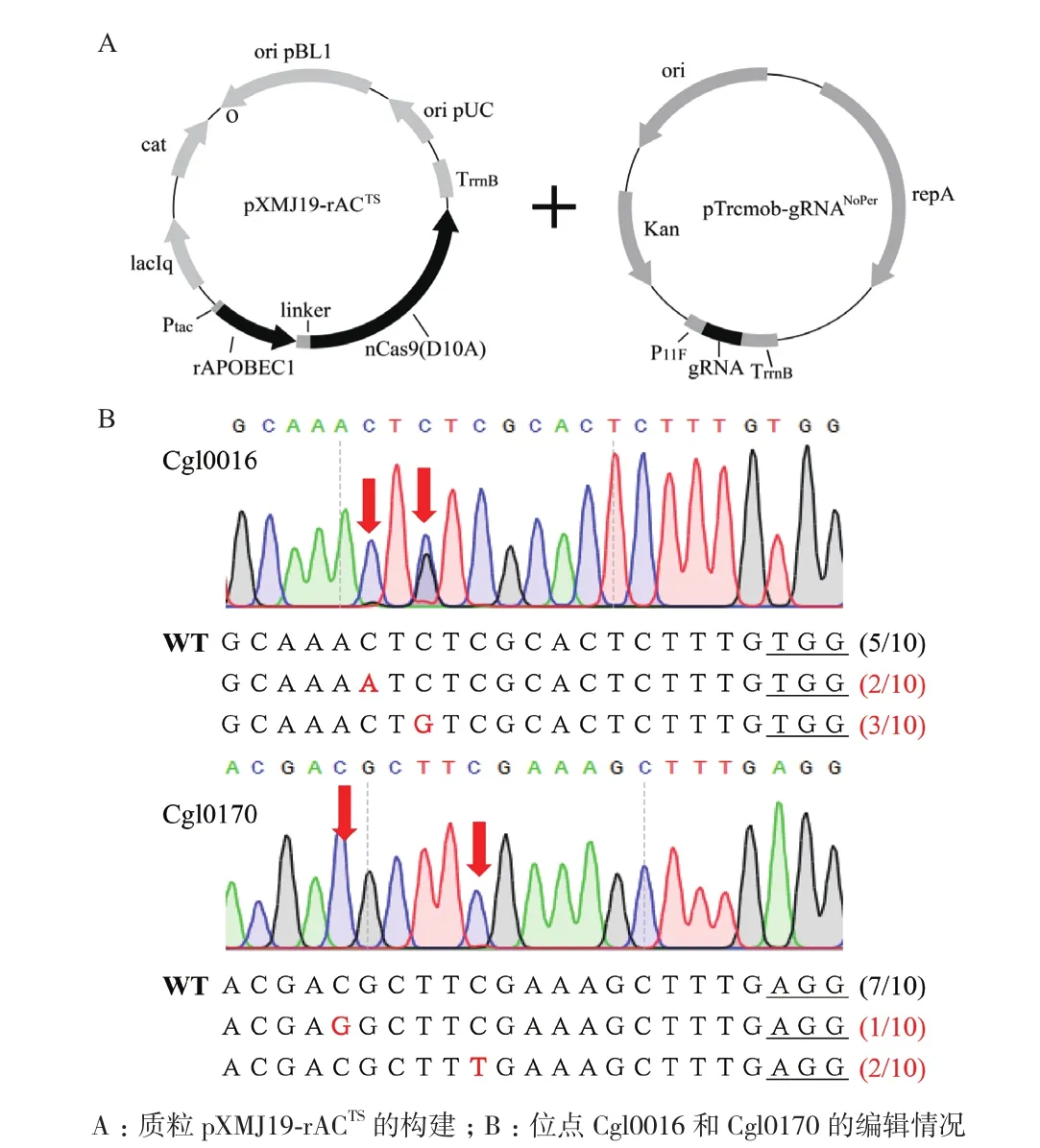
图1 质粒pXMJ19-rACTS的构建及碱基编辑结果
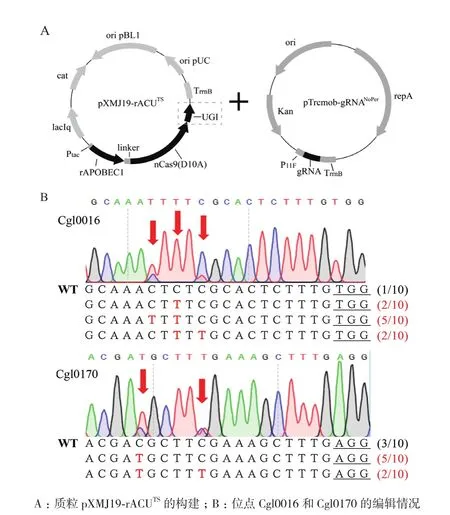
图2 质粒pXMJ19-rACUTS的构建及碱基编辑结果
2.3 优化双质粒编辑系统为单质粒编辑系统
依赖双质粒的碱基编辑系统,在谷氨酸棒杆菌转化环节以及丢失质粒环节,效率较低、操作繁琐,不利于以后高通量自动化的改造菌株。因此,为了简化操作,本研究在pXMJ19-rACUTS质粒基础上,加入gRNA转录模块,获得重组质粒pXMJ19-rACU-gRNATS(图3-A)。在gRNA模块中插入ccdB反选标签,通过Golden Gate的连接方法即可快速实现靶标gRNA序列的替换。相比较于双质粒的转化,单质粒的转化效率得到明显提高(图4)。为了研究调整为单质粒结构后,编辑效率以及编辑窗口是否会受到影响,分别构建了含Cgl0016及Cgl0170的靶标gRNA序列的单质粒编辑系统。经过诱导编辑及Sanger测序,两个位点均得到编辑,且编辑窗口未发生改变(图3-B),虽然从测序峰图及编辑比例来比较,单质粒的编辑效率出现略微的下降,但编辑效率仍然较高,其中,Cgl0016中C到T的编辑比例为80%,Cgl0170中C到T的编辑比例为60%,并且均未有C到A,C到G的编辑。该实验结果证明,单质粒编辑系统可以取代双质粒编辑系统,应用于后期的研究中。
2.4 基因组中其他位点的碱基编辑测试
为了进一步验证BE3型胞嘧啶碱基编辑器在谷氨酸棒杆菌的编辑效率,并测试该碱基编辑器编辑框的范围,我们另选取了基因组中其他4个位点(Cgl0106,Cgl1492,Cgl0221,Cgl2111)进行测试。通过分别构建含靶标gRNA序列的单质粒编辑系统、诱导编辑以及Sanger测序分析,发现4个位点同样可以实现C到T的编辑(图5),除Cgl0106位点编辑效率为40%外,其他3个位点的编辑效率均高达90%-100%,充分证明了该碱基编辑器的高效性。另外还发现,该碱基编辑器的编辑框可以覆盖PAM上游-11至-19位,相比较于哺乳动物中的编辑框(PAM上游-13到-17位),范围更大,该特性推测可能是由于物种差异所造成。编辑框的扩展,将覆盖更多的基因组靶标位点,在全基因组失活基因应用中,具有较明显的优势。

图3 质粒pXMJ19-rACU-gRNATS的构建及碱基编辑结果
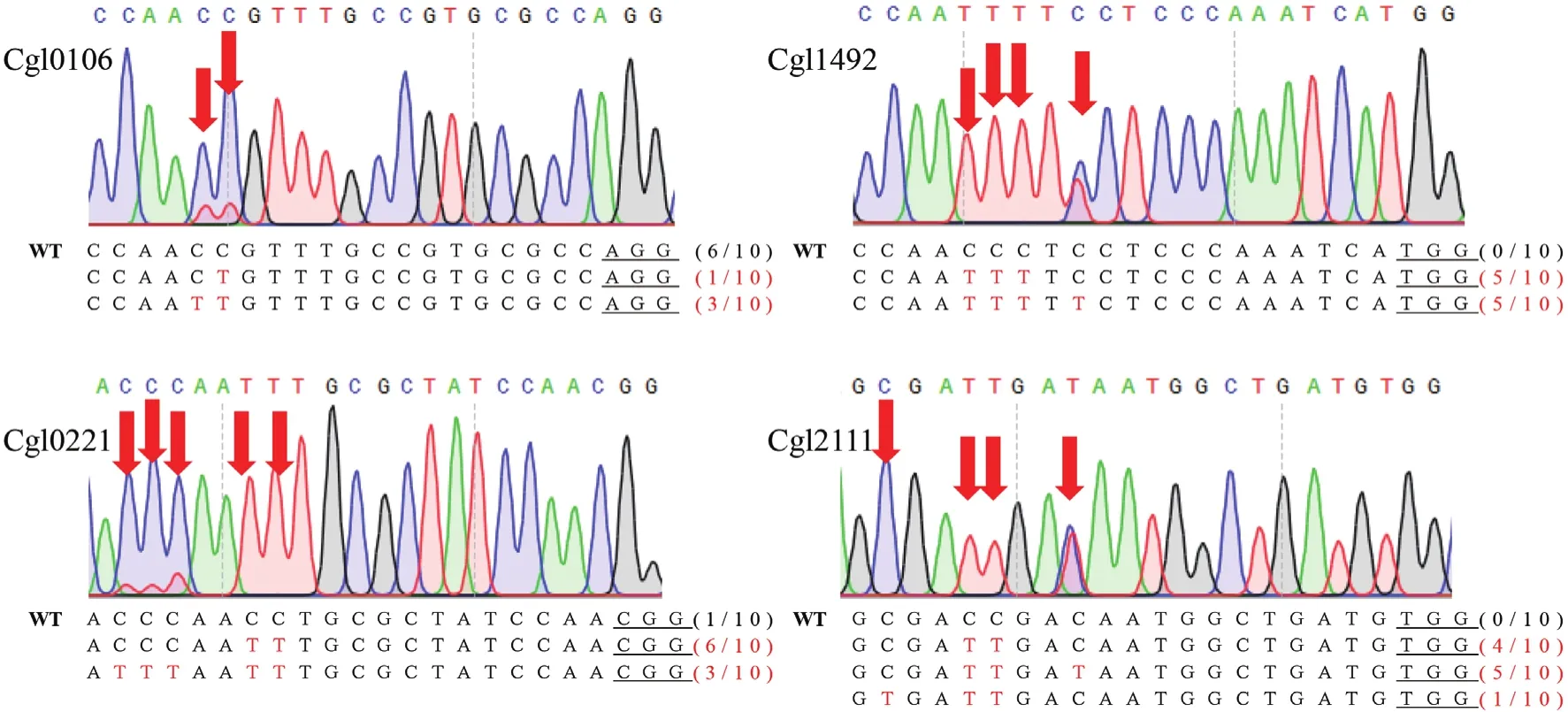
图5 谷氨酸棒杆菌基因组位点碱基编辑结果
3 讨论
谷氨酸棒杆菌是从土壤中分离出来的革兰氏阳性菌,为食品安全性菌株,在工业生产领域广泛的应用于生产各类氨基酸、有机酸等大宗化学品。近年来,随着CRISPR/Cas系统的发展与壮大,科研人员已成功在谷氨酸棒杆菌中建立了基于DSB的CRISPR/Cas9或Cpf1基因组编辑工具,为谷氨酸棒杆菌的基因组改造提供了强大的技术支持[13-14]。但是由于DSB带来的毒性、对外源模板DNA的需求,以及谷氨酸棒杆菌自身基因组的稳定导致的同源重组效率低,这些因素限制了CRISPR/Cas系统在谷氨酸棒杆菌中的应用,对于自动化高通量的基因组改造、多位点编辑等仍然存在较大难度。碱基编辑技术的兴起,为解决上述问题提供了新的研究思路。该技术结合了CRISPR/Cas系统的靶向特异性与碱基脱氨酶的催化活性,可在不产生DSB、不依赖宿主的同源重组修复且不需要外源DNA模板的情况下对基因组上位点实现高效精准的编辑,并且避免了DSB造成的靶标或非靶标序列的缺失、插入等突变。
BE3型碱基编辑器与前期已开发的碱基编辑技术MACBETH相比,主要存在以下几点不同:(1)编辑窗口不同,MACBETH编辑窗口为PAM上游-16到-20位,而BE3的编辑窗口覆盖范围较宽,为PAM上游-11到-19位;(2)编辑效率存在差异,MACBETH技术在不添加UGI蛋白的情况下,编辑效率即可达到较高,C到T的碱基编辑效率高达90%-100%,而BE3型碱基编辑器在不添加UGI蛋白时,编辑效率较低,UGI蛋白的添加能显著提高碱基编辑效率,使得C到T的碱基编辑效率同样高达90%-100%。BE3型碱基编辑器的开发能够很好的与MACBETH互补,有助于覆盖更多的基因组靶标位点,为自动化高通量的基因组改造提供更多的选择,从而加速谷氨酸棒杆菌的基础与应用研究。
4 结论
本研究首先融合表达rAPOBEC1-nCas9(D10A)蛋白,实现在谷氨酸棒杆菌中C到T的编辑,编辑比例较低(0-20%)。添加UGI蛋白后,显著提高了碱基编辑效率,使得C到T的碱基编辑效率高达90%。为了简化操作,将双质粒碱基编辑系统优化为单质粒碱基编辑系统,并且未明显影响碱基编辑效率及编辑窗口。最后对基因组中其他位点进行测试,证明编辑框为PAM上游-11到-19位,编辑效率为40%-100%。
猜你喜欢
电脑爱好者(2021年3期)2021-02-06
教学考试(高考生物)(2020年6期)2020-11-23
化工时刊(2020年8期)2020-01-12
食品与生物技术学报(2020年8期)2020-01-06
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
党的生活·党员电教与远程教育(2019年1期)2019-03-06
铁道通信信号(2018年7期)2018-08-29
发明与创新·大科技(2018年8期)2018-02-20
百科知识(2015年13期)2015-09-10
