
基于CRISPR-Cas13家族的RNA编辑系统及其最新进展
2020-03-19 09:32陈敏洁唐桂月洪香娜郝沛江静李轩
生物技术通报 2020年3期
陈敏洁 唐桂月 洪香娜 郝沛 江静 李轩
(1. 中国科学院分子植物科学卓越中心合成生物学重点实验室,上海 200032;2. 中国科学院上海巴斯德研究所病原发现与大数据中心,上海 200000;3. 河南大学生命科学学院棉花生物学国家重点实验室植物逆境生物学重点实验室,开封 475004)
RNA作为一种重要的生物大分子,参与细胞生命代谢的整个过程,其代谢异常会导致多种疾病的发生[1]。故了解RNA的功能和代谢对于人类健康至关重要。目前,靶向DNA的技术如锌指核酸酶(Zinc finger nuclease,ZFN)、转录激活因子样效应物核酸酶(Transcription activator-like effector nucleases,TALENs)、CRISPR-Cas9 或 Cas12 等[2]现已成为生物医学实验室的常规操作,并且已经初步应用于临床医学领域。然而DNA序列一旦干预成功,在很大程度上会遗传给后代,具有永久性与不可恢复性。与相对稳定遗传的DNA不同,RNA引起的改变是暂时、非永久性的。但是RNA生物学的极度复杂性导致很难精确地靶向、跟踪或编辑。目前靶向RNA的技术主要包括RNAi、MS2技术、ADARs技术及CRISPR-Cas13 技术等[3]。
本文主要对目前最新的靶向RNA技术的CRISPR-Cas13家族的分类及防御机制进行综述,介绍了 CRISPR-Cas13 技术的应用以及基于CRISPRCas13家族的RNA编辑系统的最新研究进展,并对目前CRISPR-Cas13 RNA编辑技术体系存在的问题进行分析和对未来的发展进行展望。
1 CRISPR-Cas13家族和分类
CRISPR-Cas13是基于细菌免疫系统的RNA靶向和编辑系统,可保护其免受病毒侵袭。该系统类似于CRISPR-Cas9系统,但与靶向DNA的Cas9不同,Cas13蛋白仅靶向切割单链RNA。CRISPRCas13(以前称为 C2c2)[4]是 CRISPR-Cas系统中的第二大类第VI型,是家族中目前发现的唯一只靶向ssRNA的系统。它包含单一的效应蛋白质Cas13,与 CRISPR RNA(crRNA)组装时形成一个由crRNA引导的RNA靶向效应复合物。迄今为止研究的所有Cas13蛋白都具有两种不同的核糖核酸酶活性[5],一种是RNase负责pre-crRNA的预处理形成成熟的VI型干扰复合物[6];而另一种RNase 活性由两个较高等真核生物和原核生物核苷酸结合(Higher eukaryotes and prokaryotes nuceotide-binding,HEPN)结构域提供,是target-RNA 在病毒干扰过程中降解所必需的,研究证明这两个HEPN结构域中的单点突变会完全消除 RNA 的切割[4,7-8]。HEPN 结构域能够帮助切割 ssRNA,但不能切割 dsRNA、ssDNA或 dsDNA,且当RNA 靶中存在折叠结构时,Cas13优先在非碱基配对的 ssRNA 区域进行剪切[9]。
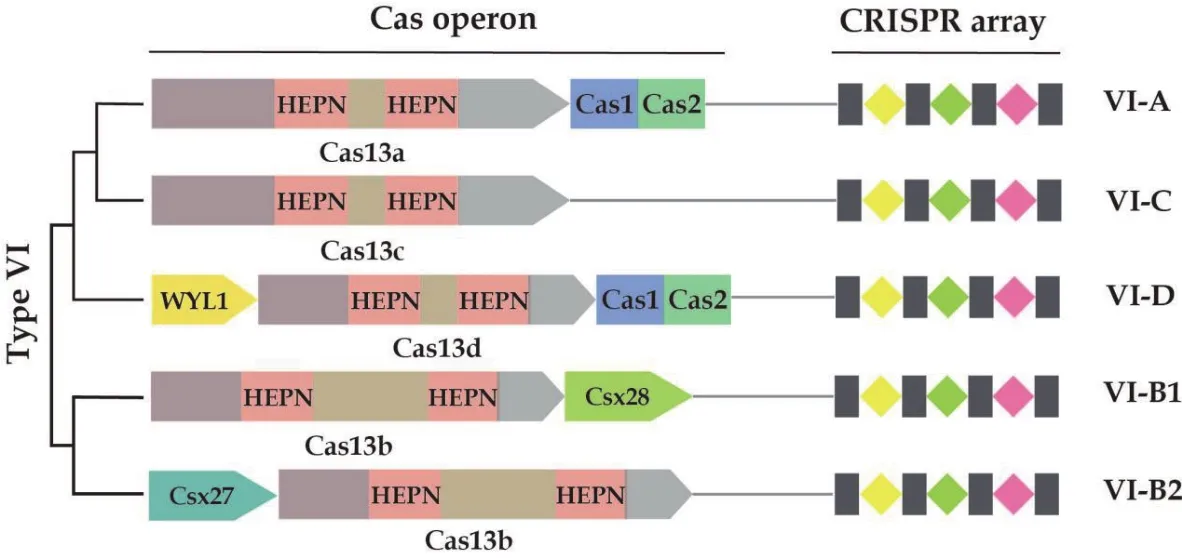
图1 CRISPR-Cas13家族分类图
目前CRISPR-Cas13根据系统发育可分为4个亚型(A、B1、B2、C 和 D)[6,10](图 1)。同源序列分析表明Cas13四个亚型具有低同源性,且同源序列仅限于HEPN结构域位点。HEPN结构域可以存在于Cas13不同位置,在VI-B型基因座中还存在其它特征,VI-B1系统具有额外的ORF,CSX28,其编码具有一个跨膜结构域(或可能是信号肽)和一个HEPN结构域的小蛋白质。然而,VI-B2系统在许多情况下(但并非所有)存在不同的ORF,Csx 27,也编码一个小蛋白质,似乎包含3-4个预测的跨膜结构域,从而进一步被划分为B1和B2亚型[11]。最新发现的亚型VI-D和相关的效应蛋白 Cas13d,它们主要存在于Eubacterium和Ruminococcus两种细菌中[6,11]。VI-D亚型比以前发现的所有亚型都小得多(比先前鉴定的Cas13蛋白小约26%)[6]。此外,与VI-B系统一样,绝大多数VI-D系统都含有相关的辅助蛋白,这些蛋白质都含有WYL结构域,这个结构域通常与原核防御系统有关[12]。除了系统发育特征外,亚型之间的功能也存在差异。例如,Seletsky等[13]对11个Cas13a 同系物的pre-crRNA 处理行为进行的研究。
2 CRISPR-Cas13家族防御的分子机制
CRISPR-Cas13系统的防御过程与其他CRISPRCas系统相似,包括 3 个阶段:第一是新间隔序列的获取[14],称适应阶段;第二是 crRNA的合成[15],即 CRISPR基因座的表达;第三是对外来核酸的抵御,称干扰阶段[16](图2)。适应阶段获取外源核酸序列整合在CRISPR阵列中。CRISPR阵列包括交替的半回文直接重复序列和由整合酶Cas蛋白插入的外源核酸“间隔”序列组成。一直以来Cas1、Cas2被认为是参与新间隔获取的主要功能蛋白[17],然而在一些情况下,VI型CRISPR-Cas缺乏适应基因(Cas1、Cas2)[11],表明它们可能利用同一基因组中其他 CRISPR-Cas基因座的适应模块,或者失去了它们的适应模块,不再积极地获得新的间隔[18]。
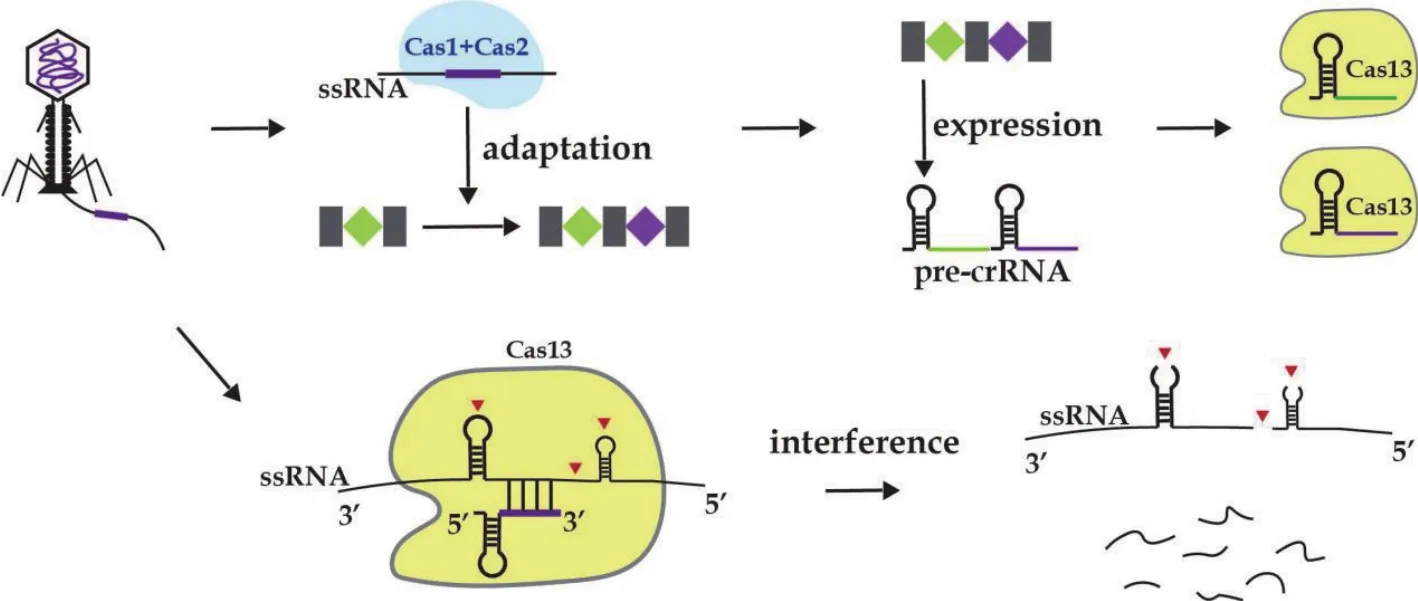
图2 CRISPR-Cas13家族分子防御过程
CRISPR基因座表达起始产物pre-crRNA,经过相关Cas 蛋白和RNA 核酸酶的加工形成成熟crRNA,此过程即为 CRISPR 基因座的表达[13,19]。研究表明,VI型crRNA处理是由Cas13中的一种专用RNA核酸酶进行的。East-Seletsky等[5]首先证明Cas13a具有pre-crRNA加工活性,并且该活性不需要HEPN核酸酶结构域。Cas13b和Cas13d也被证明具有pre-crRNA处理活性,在Cas13b的系统中,pre-crRNA被加工成一个66 nt的成熟crRNA(一个30 nt的5' -间隔和一个36 nt的3'-直接重复序列)[11]。而Cas13d pre-RNA加工产生的成熟crRNA具有与Cas13a家族相似的重复间隔结构[6]。鉴于在 VI-A、B和D型系统中pre-crRNA处理的保守性,Cas13c也很可能保持切割pre-crRNA的能力,但还需要进一步研究证实。
成熟的crRNA能够招募Cas13蛋白,从而完成防御入侵最重要的一步干扰阶段,准确识别外源核酸并对其进行精准剪切。CRISPR-Cas13的4个亚型中,形成的crRNA结构存在一定的差别。在Cas13a:crRNA复合物中,直接重复序列形成5-6个Watson-Crick碱基对的茎,由保守的2nt凸起(AC或AA)破坏,依赖于Cas13a一个7-9nt的茎环区域[9]。虽然目前还没有高分辨率的VI-B型crRNA结构,但RNA二级结构分析表明,与VI-A、VI-C和VI-D crRNA相比,VI-B crRNAs具有更长的直接重复序列茎(总共9-14 bp),通常具有几个不成对的区域和凸起,因此茎环更小[20]。除茎环结构之外,在 VI-B 型系统中,直接重复序列相对于间隔的顺序也不同,直接重复序列的茎环位于间隔的3'处,而在VI-A、VI-C和VI-D系统中观察到的直接重复序列在间隔的5'处结构不同。与VI-A型和VI-B型crRNA 相比,VI-C型crRNAs直接重复序列的长度较短(~30 nt)[21],表明它在存在不同的适应模块时可能与VI-A、B和D系统共同进化。最后VI-D型,包含具有36 nt 直接重复序列的crRNA,形成了一个保守的8-10 nt茎,带有一个4-6 nt的环,一个5-10 nt的5'侧翼单链区(在pre-crRNA中)和一个包含保守的末端AAAAC基序的5-7 nt的3'侧翼单链区[6,12]。
Cas13:crRNA核酸靶向复合物与靶标核酸序列碱基互补帮助完成搜索过程和初始杂交。除此之外,如Cas9干扰复合物需要检测dsDNA靶位点侧翼的原间隔子相邻基序(PAM)序列的存在也会帮助识别靶标序列[22-23]。然而,Abudayyeh等[4]探索了Cas13a对于在每个crRNA靶位点侧翼的核苷酸是否表现出相同的序列偏好。他们发现对于LshCas13a,protospacer-3'侧翼的第一个核苷酸表现出对A、U或C(H)而不是G的偏好性。通过体外RNA降解实验证实了该结果,结果表明该位置存在G核苷酸会显着降低HEPN核酸酶的活性,而A、U和C会导致最大活性[24]。为了避免与PAM在“自我”CRISPR基因组学中所定义混淆,作者决定将这一序列偏好称为“Protospacer flanking site”(PFS),而不是“Protospacer adjacent motif”(PAM)。自从首次报道PFS 偏好LshCas13a 以来,PFS 偏好也被证实存在于其他几种 Cas13酶中。使用体外裂解试验,最初证明LwaCas13a具有轻微的“ H” PFS偏好[25]。此作者后来又证明,当对细菌筛选[26]或人类细胞系质粒文库筛选[20]PFS时,LwaCas13a不会表现出任何可检测到的PFS偏好,并且靶向人细胞中含ssRNA的G-PFS不会导致任何缺陷定位效率。
Cas13b酶的PFS要求也有所不同。在细菌中对于来自Bergeyella zoohelcum和Prevotella buccae菌的两个不同的Cas13b靶标文库筛选时,D(A、U或G)5'PFS和NAN或NNA 3'PFS是实现最佳RNA靶向所必需的[11]。相反,当对来自PspCas13b进行细菌中错配的目标文库实验时发现,有效的靶标RNA裂解不需要优选的PFS[20]。与Cas13a或Cas13b相反,对于迄今为止研究的任何Cas13d直系同源物,使用细菌筛选策略均未检测到PFS序列。目前,关于Cas13c酶家族是否存在PFS偏好尚无定论。
3 CRISPR-Cas13分子技术的应用
目前利用CRISPR-Cas13系统可对RNA进行编辑、敲除、检测、追踪及成像等[3]。Cas13a是第一个被用于靶向RNA切割的效应蛋白。Abudayyeh等[4]通过异源表达的Cas13a可以保护大肠杆菌免受MS2噬菌体的侵入,并首次在大肠杆菌中实现RNA敲除。作者通过改用Leptotrichia wadei(Lwa)来源的Cas13a实现了在哺乳动物细胞中进行RNA敲除[26]。基于Cas13a的RNA敲除系统与传统的RNAi技术相比,RNA敲除能力相当但Cas13a脱靶率低且可以靶向细胞核内RNA。Cas13a的RNA敲除技术目前已经被应用到许多领域,如植物病毒敲除—芜菁花叶病毒(TuMV,一种RNA病毒)[27-28],马铃薯Y病毒(PVY)等[29];哺乳动物细胞的单链RNA病毒敲除—淋巴细胞性脑膜炎病毒(LCMV),甲型流感病毒(IAV),水泡性口炎病毒(VSV)等[30],癌症相关的突变基因敲除,如KRAS等[31]。Cas13a被激活后附带的非特异性RNA酶切活性目前还被应用于核酸快速检测,如 Gootenberg等[25,32-34]开发的一种 “SHERLOCK”检测系统可以在短时间内实现从患者体液样品中进行无仪器的病毒的检测。此外,Abudayyeh等[26]将生物素与dLwaCas13a融合的用于RNA免疫沉淀,通过将dLwaCas13a与负反馈(NF)系统结合使用,还可用于RNA成像的dLwaCas13a-NF系统。
Cas13d作为目前已知最短的Cas13蛋白,使其成为进一步开发靶向RNA 工具的潜在平台[6,12,35]。Yan 等[12]与 Konermann 等[6]将不同的 Cas13d 与不同的融合标签组合,实现在人细胞中的RNA敲除。此外,Cas13d目前还被应用于活细胞RNA成像[36]。Cas13d平均大小为930 aa,是哺乳动物细胞中表征的最小的2类CRISPR效应子,这使得Cas13d结构域可以与编码多个gRNA的CRISPR阵列配对。同时,Cas13d基因组长度符合慢病毒递送载体的包装尺寸,Cas13d-慢病毒载体将成为原代细胞和体内递送的有力工具。
4 基于CRISPR-Cas13的RNA碱基精确编辑系统
4.1 基于CRISPR-Cas13的RNA碱基A>I精准编辑系统
靶向RNA编辑作为最新的研究方法,其不可永久性遗传为治疗某些遗传性疾病提供一种更安全,更有效的替代方法。Cox等[20]将VI型CRISPRCas系统中无催化活性的dPspCas13b与ADAR2融合 的 REPAIR(RNA Editing for Programmable A-to-I Replacement)RNA碱基编辑系统在哺乳动物细胞中成功编辑与疾病相关的基因。REPAIR系统利用dcas13b指导ADAR2定点编辑最重要的两个部分:具有催化活性增强的突变体ADAR2dd;特异性识别目标A·C错配碱基。ADAR2是一种广泛存在于哺乳动物之中的腺苷脱氨酶,可对特异性的双链RNA进行A(腺苷)>I(肌苷)脱氨,肌苷被当成鸟嘌呤,并与胞嘧啶配对,从而实现碱基A>G的转变[37-38]。REPAIR系统目前主要分为两个版本。REPAIRv1系统对靶序列具有较好的兼容性,在靶向荧光素Gluc RNA时,对靶目标16种可能的基序都检测到编辑。REPAIRv1系统靶向Gluc的编辑效率可达89%,具有较高的编辑效率,但在靶向内源基因PPIB编辑率只有28%。为此作者在Cas13b与ADAR2之间加上一个多肽,结果REPAIRv1系统编辑效率提高,说明Cas13与ADAR2之间的连接多肽在改善系统编辑率中起到一定的作用。REPAIRv1系统相比于其他传统RNA编辑技术—以ADAR2原始底物的编辑系统(如 BoxB- ADAR2[39]、全长 ADAR2[40])编辑率高,但最近报导的以ADAR2原始底物的最新编辑系统的编辑率与REPAIRv1相当[41]。相比脱靶率,转录组测序显示REPAIRv1的脱靶编辑率与BoxB-ADAR相当,但大于全长ADAR2的脱靶率,且脱靶位点主要集中在ADAR2原始底物结构基序部分。REPAIRv1脱靶效应主要是由于ADAR2的底物偏好性以及ADAR2dd的过度表达而引起。为了提高REPAIR的特异性,Cox等[20]通过在ADAR2上引入新突变(ADAR2 DD -E488Q-T375G)形成REPAIRv2系统,该系统在靶向Gluc其脱靶率低于REPAIRv1系统的约900倍,但编辑率也随之降低(表1)。

表1 基于CRISPR-Cas13的RNA编辑系统编辑率与脱靶率
此外,本课题组利用无活性的dCas13a与ADAR2融合实现在酵母中的定点编辑[42](图3)。利用结合特异序列单链RNA能力的Cas13a,首次在模式生物裂殖酵母(S. pombe)中实现了靶向内源RNA和降解靶标RNA的功能。进一步,再利用丧失RNA切割活性的突变dCas13a与人源的RNA腺嘌呤脱氨酶催化结构域(hADAR2d)融合,引入到裂殖酵母中;再通过设计指导RNA,实现了对内源RNA的精确定点编辑。对RNA定点编辑系统的参数(编辑碱基位置、距离、指导RNA结构和长度等)进行优化,优化后的编辑系统对内源靶标RNA的编辑效率达到了59%。
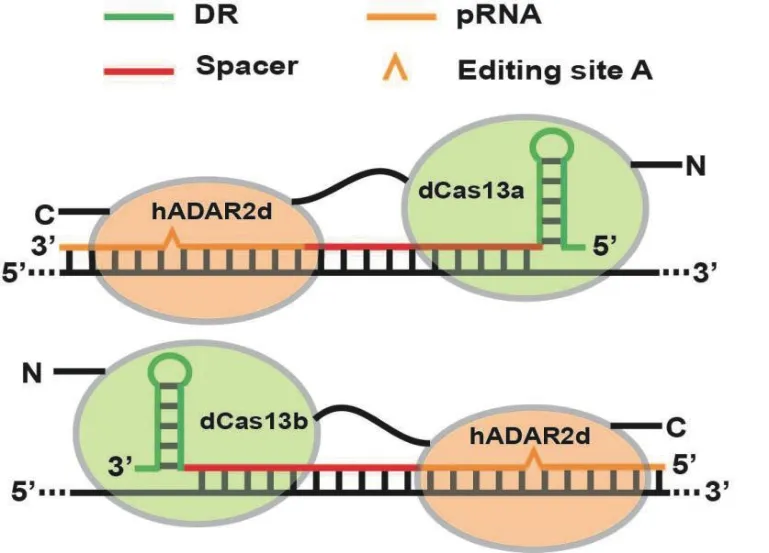
图3 dCas13a与dCas13b介导的RNA碱基编辑系统的比较[42]
4.2 基于CRISPR-Cas13的RNA碱基C>U精准编辑系统
Abudayyeh等[43]最近发表的一篇关于对RNA实现胞苷(C)>尿苷(U)的精确编辑系统RESCUE(RNA Editing for Specific C-to-U Exchange),扩展RNA编辑的功能,扩大了可解决的疾病突变和蛋白质修饰的范围。
RESCUE系统利用无催化活性的靶向RNA的CRISPR-Cas13b(dCas13b)与改造后具有 C>U 的ADAR2的腺嘌呤脱氨酶域融合而成。ADAR2具有天然A>I脱氨活性,作者根据ADAR2与底物结合的结构初步突变了与底物结合的3个关键位点(V351G、S486A、T375S)使其具有C>U的脱氨作用,并在酵母中靶向外源基因Gluc具有15%的编辑率,作者在此基础之上又经过16轮突变筛选获得具有较高编辑效率的RESCUE16系统。RESCUE系统在靶向16种Gluc的靶基序均具有编辑,并且gRNA spacer具有C或U错配时具有较高的编辑率。RESCUE在靶向HEK293细胞内源基因时,编辑最高能达到42%左右,其偏好性主要集中在5' U或A且受U或者 A 错配位置影响。此外,作者还在体外证明了该系统对DNA、sRNA没有胞苷脱氨作用。为了证明该系统可应用于治疗作用,作者利用RESCUE系统调节STAT和WNT-β-catenin信号通路。将具有RESCUE质粒和gRNA质粒转染HEK293FT细胞和人脐静脉内皮细胞(HUVECs),通过靶向 β-catenin上CTNNB1的已知磷酸化残基,其编辑率最高可达28%,而被激活WNT-β-catenin信号传导最高可以提高到5倍。
改造后的ADAR2仍具有A>I的活性,Cas13b能够将pre-crRNA加工形成A>I/C>I的gRNA同时实现A>I和C>U的编辑。作者通过优化gRNA,可同时靶向HEK293FT细胞中CTNNB1A>I和C>U的编辑。RESCUE在靶向Gluc引起A>I脱靶位点有1695个,C>U脱靶位点具有188个。为此作者又通过引进新的突变体,并发现S375A、S375C、S375N、N473I的引起脱靶率大幅度下降,其中S375A A>I脱靶率降到139,C>U 脱靶率降到103且编辑率没有太大影响。
RNA编辑的一个主要优点是它的可逆性,相比之下,DNA水平上的变化是永久性的。RESCUE能够被gRNA引导至目标RNA,实现C>U编辑。该系统不仅扩大了RNA编辑系统的范围,并为RNA编辑技术的潜在临床应用奠定了基础[44],同时利用RESCUE扩展靶向能力意味着编辑新靶标的到来,通过磷酸化、甲基化和糖基化等蛋白翻译后修饰可以调节蛋白的活性和功能的位点如今都可作为编辑的新靶标。但RESCUE仍具有较高的A>I的RNA编辑活性,改造编辑酶或优化其他参数系统减少脱靶率仍是目前需解决的问题。
5 展望
RNA是细胞中最重要的信使分子之一。RNA的功能和动态代谢的表征对于理解生命过程至关重要。然而RNA是快速合成、代谢使得在体内很难靶向,跟踪或编辑,CRISPR-Cas13系统为该领域提供了新的思路。基于Cas13蛋白开发的RNA工具在疾病研究和临床治疗中的应用将为人类医疗保健的巨大进步作出贡献。
截至目前,人类致病突变最多的一类是点突变(也称为单核苷酸多态性)[47]。基于CRISPR的靶向DNA 的碱基编辑系统—CBEs(C>U)、ABEs(A>U)基本上能够实现4种碱基突变(C >T、A > G、T>C、G>A)的修复,但靶向DNA具有可遗传性和不可修复性等风险。相反,RNA编辑系统可以从RNA水平上进行修复而不会永久性地遗传(表2)。目前基于CRISPR-Cas13家族的编辑系统A>I(REPAIRv1)以及C>U(RESCUE)基本上能修复致病突变的点突变。通过合理的设计方法,如gRNA的优化以及编辑蛋白的定向进化,可以进一步提高系统的特异性和效率。尽管目前基于RNA碱基编辑系统的例子非常令人鼓舞,但是将大蛋白递送到特定组织、体内脱靶点突变的潜在生物学后果等相关工作依然具有挑战性。因此,新型RNA碱基编辑器传送系统的开发,包括针对特定组织的系统,可能是未来几年的主要重点工作。
猜你喜欢
世界科学技术-中医药现代化(2022年9期)2023-01-17
湖北农业科学(2022年11期)2022-07-18
军民两用技术与产品(2021年10期)2021-03-16
教学考试(高考生物)(2020年6期)2020-11-23
生物信息学(2020年1期)2020-05-16
食品与生物技术学报(2020年8期)2020-01-06
世界农药(2019年3期)2019-09-10
学苑创造·B版(2019年5期)2019-06-14
科学24小时(2019年5期)2019-06-11
天津科技大学学报(2016年1期)2016-02-28
